Journal of Proteomics ( IF 3.3 ) Pub Date : 2020-08-03 , DOI: 10.1016/j.jprot.2020.103921 Martin G Klatt 1 , Zita E H Aretz 2 , Michael Curcio 1 , Ron S Gejman 3 , Heather F Jones 4 , David A Scheinberg 4
|
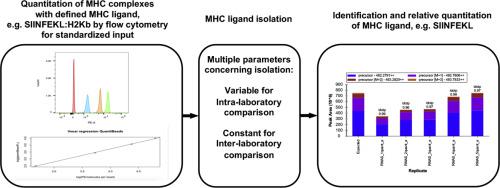
Characterization of MHC-bound peptides by mass spectrometry (MS) is an essential technique for immunologic studies. Many efforts have been made to quantify the number of MHC-presented ligands by MS and to define the limits of detection of a specific MHC ligand. However, these experiments are often complex and comparisons across different laboratories are challenging. Therefore, we compared and orthogonally validated quantitation of peptide:MHC complexes by radioimmunoassay and flow cytometry using TCR mimic antibodies in three model systems to establish a method to control the experimental input of peptide MHC:complexes for MS analysis. Following isolation of MHC-bound peptides we identified and quantified an MHC ligand of interest with high correlation to the initial input. We found that the diversity of the presented ligandome, as well as the peptide sequence itself affected the detection of the target peptide. Furthermore, results were applicable from these model systems to unmodified cell lines with a tight correlation between HLA-A*02 complex input and the number of identified HLA-A*02 ligands. Overall, this framework provides an easily accessible experimental setup that offers the opportunity to control the peptide:MHC input and in this way compare immunopeptidome experiments not only within but also between laboratories, independent of their experimental approach.
Significance
Although immunopeptidomics is an essential tool for the characterization of MHCbound peptides on the cell surface, there are no easily applicable established protocols available that allow comparison of immunopeptidome experiments across laboratories. Here, we demonstrate that controlling the peptide:MHC input for immunopurification and LC-MS/MS experiments by flow cytometry in pre-defined model systems allows the generation of qualitative and quantitative data that can easily be compared between investigators, independently of their methods for MHC ligand isolation for MS.
中文翻译:
用于识别 MHC 结合肽的输入控制模型系统,可实现免疫肽组实验的实验室比较。
通过质谱法 (MS) 表征 MHC 结合的肽是免疫学研究的一项基本技术。已经做出了许多努力来量化由 MS 提供的 MHC 配体的数量,并定义特定 MHC 配体的检测限。然而,这些实验通常很复杂,不同实验室之间的比较具有挑战性。因此,我们在三个模型系统中使用 TCR 模拟抗体通过放射免疫测定和流式细胞术比较和正交验证肽:MHC 复合物的定量,以建立一种方法来控制肽 MHC:复合物的实验输入,用于 MS 分析。在分离 MHC 结合肽后,我们确定并量化了与初始输入高度相关的目标 MHC 配体。我们发现所呈现的配体组的多样性,以及肽序列本身影响目标肽的检测。此外,这些模型系统的结果适用于未修饰的细胞系,在 HLA-A*02 复合物输入与已识别的 HLA-A*02 配体数量之间存在紧密相关性。总体而言,该框架提供了一个易于访问的实验设置,提供了控制肽:MHC 输入的机会,并以这种方式不仅在实验室内而且在实验室之间比较免疫肽组实验,独立于他们的实验方法。
意义
尽管免疫肽组学是表征细胞表面 MHC 结合肽的重要工具,但尚无易于应用的既定协议,可用于比较实验室间的免疫肽组实验。在这里,我们证明了在预定义的模型系统中通过流式细胞术控制用于免疫纯化和 LC-MS/MS 实验的肽:MHC 输入允许生成定性和定量数据,这些数据可以很容易地在研究人员之间进行比较,而与他们的方法无关。用于 MS 的 MHC 配体分离。

























 京公网安备 11010802027423号
京公网安备 11010802027423号