Biochimie ( IF 3.9 ) Pub Date : 2020-07-25 , DOI: 10.1016/j.biochi.2020.07.010 Qiuhong Liu 1 , Jinqiao Zhou 2 , Jing Gao 1 , Wentao Ma 1 , Shilei Wang 1 , Lihua Xing 1
|
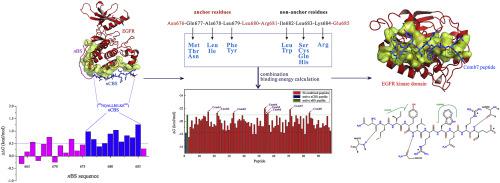
Although a variety of tyrosine kinase inhibitors (TKIs) have been developed to target human epidermal growth factor receptor (EGFR) for lung cancer therapy, many patients treated with first-line small-molecule TKIs are clinically observed to eventually establish drug-resistant mutations T790 M and C797S around kinase active site, which play a primary role in development of acquired drug resistance to first-generation reversible and second-generation irreversible TKIs, respectively. Here, instead of developing small-molecule drugs to directly target the active site, we attempt to derive self-inhibitory peptides (SIPs) from the EGFR:EGFR asymmetric dimerization interface, where is separated from kinase active site and has a relatively low conservation as compared to the active site. It is found that the dimerization is a typical peptide-mediated protein interaction, where the first EGFR N-lobe adopts an N-terminal binding sequence (nBS) to interact with the dimerization interface of second EGFR C-lobe. A core binding sequence (nCBS, 676NQALLRILKE68) is identified in the nBS region as hotspot segment, which is then rationally optimized to generate a number of SIPs. Consequently, three designed SIPs are determined as promising candidates; they have high affinity to EGFR kinase domain, strong competitive potency with native nBS peptide for the dimerization interface, and effective cytotoxicity on human lung cancer cell lines. It is also demonstrated that these peptides are insensitive to drug-resistant EGFR T790 M/C797S mutation at molecular and cellular levels, and exhibit a good selectivity for EGFR over HER2 at molecular level.
中文翻译:
EGFR二聚化破坏肽的合理设计:在靶向肺癌治疗中对抗耐药性的新策略。
尽管已开发出多种酪氨酸激酶抑制剂(TKI)靶向人类表皮生长因子受体(EGFR)进行肺癌治疗,但临床上观察到许多用一线小分子TKI治疗的患者最终建立了耐药突变T790激酶活性位点附近的M和C797S,分别在第一代可逆和第二代不可逆TKI的获得性耐药发展中起主要作用。在这里,我们没有开发直接靶向活性位点的小分子药物,而是尝试从EGFR:EGFR不对称二聚化界面衍生自抑制肽(SIP),该界面与激酶活性位点分开并且保守性相对较低。与活动网站相比n BS)与第二个EGFR C叶的二聚化界面相互作用。在n BS区域中将核心绑定序列(n CBS,676 NQALLRILKE 68)标识为热点片段,然后对其进行合理优化以生成多个SIP。因此,三个设计好的SIP被确定为有前途的候选者。它们对EGFR激酶结构域具有高亲和力,与天然n具有很强的竞争能力BS肽具有二聚化作用,对人肺癌细胞系具有有效的细胞毒性。还证明这些肽在分子和细胞水平上对耐药的EGFR T790 M / C797S突变不敏感,并且在分子水平上对HER2表现出对EGFR的良好选择性。


























 京公网安备 11010802027423号
京公网安备 11010802027423号