Journal of Molecular Graphics and Modelling ( IF 2.9 ) Pub Date : 2020-07-19 , DOI: 10.1016/j.jmgm.2020.107677 Elżbieta Dziadyk 1 , Janusz Trawczyński 1 , Bartłomiej M Szyja 1
|
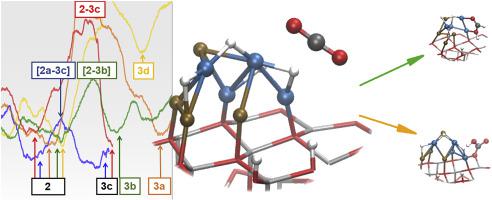
Metal nanoparticles supported on semiconductor surfaces have been proposed as promising nanocatalyst candidates of conversion to energy carrier molecules such as formic acid or carbon monoxide, which can be used as a feedstock for fuels synthesis. This study is focused on the bimetallic Cu/Ni nanoparticles supported on the ZnO. The respective reaction mechanisms have been studied by means of the Molecular Dynamics with the DFT methodology. The results suggest that on CuNi/ZnO hydrogenation to formate pathway is more favorable than carboxyl route. These pathways are competitive with the reduction to CO.
中文翻译:
DFT分子动力学模拟研究NiCu / ZnO催化CO2加氢的途径。
已经提出了负载在半导体表面上的金属纳米颗粒作为有希望的纳米催化剂候选物。 转化为能量载体分子,例如甲酸或一氧化碳,可用作分子合成燃料。这项研究的重点是负载在ZnO上的双金属Cu / Ni纳米颗粒。已经通过分子动力学和DFT方法研究了各自的反应机理。结果表明,在CuNi / ZnO上氢化成甲酸途径比羧基途径更有利。这些途径与 减少到CO。

























 京公网安备 11010802027423号
京公网安备 11010802027423号