当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Extending scaled-interaction adaptive-partitioning QM/MM to covalently bonded systems.
Physical Chemistry Chemical Physics ( IF 3.3 ) Pub Date : 2020-07-10 , DOI: 10.1039/d0cp02855j Zeng-Hui Yang 1
Physical Chemistry Chemical Physics ( IF 3.3 ) Pub Date : 2020-07-10 , DOI: 10.1039/d0cp02855j Zeng-Hui Yang 1
Affiliation
|
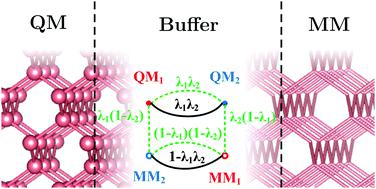
Quantum mechanics/molecular mechanics (QM/MM) is the method of choice for atomistic simulations of large systems that can be partitioned into active and environmental regions. Adaptive-partitioning (AP) methods extend the applicability of QM/MM, allowing active regions to change during the simulation. AP methods achieve continuous potential energy surface (PES) by introducing buffer regions in which atoms have both QM and MM characters. Most of the existing AP-QM/MM methods require multiple QM calculations per time step, which can be expensive for systems with many atoms in buffer regions. Although one can lower the computational cost by grouping atoms into fragments, this may not be possible for all systems, especially for applications in covalent solids. The SISPA method [Field, J. Chem. Theory Comput., 2017, 13, 2342] differs from other AP-QM/MM methods by only requiring one QM calculation per time step, but it has the flaw that the QM charge density and wavefunction near the buffer/MM boundary tend to those of isolated atoms/fragments. Besides, regular QM/MM methods for treating covalent bonds cut by the QM/MM boundary are incompatible with SISPA. Due to these flaws, SISPA in its original form cannot treat covalently bonded systems properly. In this work, I show that a simple modification to the SISPA method improves the treatment of covalently bonded systems. I also study the effect of correcting the charge density in SISPA by developing a density-corrected pre-scaled algorithm. I demonstrate the methods with simple molecules and bulk solids.
中文翻译:

将比例交互自适应分区QM / MM扩展到共价键合系统。
量子力学/分子力学(QM / MM)是大型系统原子模拟的一种选择方法,该系统可以划分为活动区域和环境区域。自适应分区(AP)方法扩展了QM / MM的适用范围,允许在仿真过程中更改活动区域。AP方法通过引入其中原子同时具有QM和MM特性的缓冲区来获得连续的势能面(PES)。大多数现有的AP-QM / MM方法每个时间步都需要多次QM计算,这对于缓冲区中有许多原子的系统而言可能是昂贵的。尽管可以通过将原子分组为多个片段来降低计算成本,但这并非对所有系统都可行,尤其是对于共价固体中的应用而言。SISPA方法[Field ,J.Chem.Soc。理论计算。,2017,13,2342]从其它AP-QM / MM方法相差仅需要每时间步一个QM的计算,但它有缺陷,该QM电荷密度和波函数的缓冲近/ MM边界趋于那些分离的原子的/片段。此外,常规的QM / MM方法处理被QM / MM边界切割的共价键与SISPA不兼容。由于这些缺陷,原始形式的SISPA无法正确处理共价键合的系统。在这项工作中,我证明了对SISPA方法的简单修改可以改善对共价键合体系的处理。我还研究了通过开发密度校正的预缩放算法来校正SISPA中电荷密度的效果。我演示了使用简单分子和大块固体的方法。
更新日期:2020-08-25
中文翻译:
将比例交互自适应分区QM / MM扩展到共价键合系统。
量子力学/分子力学(QM / MM)是大型系统原子模拟的一种选择方法,该系统可以划分为活动区域和环境区域。自适应分区(AP)方法扩展了QM / MM的适用范围,允许在仿真过程中更改活动区域。AP方法通过引入其中原子同时具有QM和MM特性的缓冲区来获得连续的势能面(PES)。大多数现有的AP-QM / MM方法每个时间步都需要多次QM计算,这对于缓冲区中有许多原子的系统而言可能是昂贵的。尽管可以通过将原子分组为多个片段来降低计算成本,但这并非对所有系统都可行,尤其是对于共价固体中的应用而言。SISPA方法[Field ,J.Chem.Soc。理论计算。,2017,13,2342]从其它AP-QM / MM方法相差仅需要每时间步一个QM的计算,但它有缺陷,该QM电荷密度和波函数的缓冲近/ MM边界趋于那些分离的原子的/片段。此外,常规的QM / MM方法处理被QM / MM边界切割的共价键与SISPA不兼容。由于这些缺陷,原始形式的SISPA无法正确处理共价键合的系统。在这项工作中,我证明了对SISPA方法的简单修改可以改善对共价键合体系的处理。我还研究了通过开发密度校正的预缩放算法来校正SISPA中电荷密度的效果。我演示了使用简单分子和大块固体的方法。

























 京公网安备 11010802027423号
京公网安备 11010802027423号