当前位置:
X-MOL 学术
›
ACS Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
How Noninnocent Spectator Species Improve the Oxygen Reduction Activity of Single-Atom Catalysts: Microkinetic Models from First-Principles Calculations
ACS Catalysis ( IF 12.9 ) Pub Date : 2020-07-10 , DOI: 10.1021/acscatal.0c01642 Michael Rebarchik 1 , Saurabh Bhandari 1 , Thomas Kropp 1 , Manos Mavrikakis 1
ACS Catalysis ( IF 12.9 ) Pub Date : 2020-07-10 , DOI: 10.1021/acscatal.0c01642 Michael Rebarchik 1 , Saurabh Bhandari 1 , Thomas Kropp 1 , Manos Mavrikakis 1
Affiliation
|
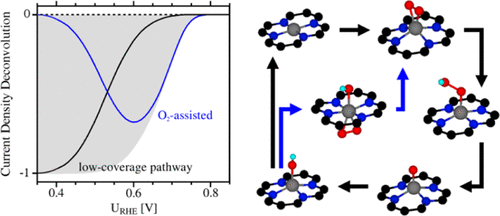
Graphene-based single-atom catalysts are promising alternatives to platinum-based catalysts for fuel cell applications. Different transition metals have been screened using electronic structure methods by estimating onset potentials from the most endergonic elementary reaction step. We calculate onset potentials for the oxygen reduction reaction on metal atoms embedded in N-substituted graphene di-vacancies by virtue of first-principles-informed microkinetic analysis. We find that for more oxophilic metals (Cr, Fe, Mn, and Ru), purely thermodynamic models systematically underestimate onset potentials. Furthermore, the oxophilic metals (Cr, Fe, Mn, and Ru) are oxidized under reaction conditions, leading to an increase in activity compared to their reduced state. Importantly, coadsorbed OmHn species actively participate in the reaction, which requires a dynamic treatment of spectator species. These findings highlight the limitations of thermodynamic analyses for electrocatalytic processes, which commonly assume the same oxidation state for each metal, and show that deviations between computational and experimental onset potentials cannot be solely attributed to the shortcomings of the electronic structure methods.
中文翻译:

非无辜的观众物种如何提高单原子催化剂的氧还原活性:第一性原理计算的微动力学模型
石墨烯基单原子催化剂是燃料电池应用中铂基催化剂的有前途的替代品。已经通过电子结构方法通过估计来自最强电子元素反应步骤的起始电位来筛选出不同的过渡金属。我们通过第一性原理的微观动力学分析计算了嵌入N-取代石墨烯双空位中的金属原子上的氧还原反应的起始电位。我们发现,对于更多的亲热金属(Cr,Fe,Mn和Ru),纯热力学模型系统地低估了起始电位。此外,亲氧金属(Cr,Fe,Mn和Ru)在反应条件下被氧化,与其还原态相比,导致活性增加。重要的是,共吸附的O m H n物种积极参与反应,这需要对观赏物种进行动态处理。这些发现凸显了电催化过程的热力学分析的局限性,通常对每种金属都假定相同的氧化态,并表明计算电位和实验起始电位之间的偏差不能仅归因于电子结构方法的缺点。
更新日期:2020-08-21
中文翻译:
非无辜的观众物种如何提高单原子催化剂的氧还原活性:第一性原理计算的微动力学模型
石墨烯基单原子催化剂是燃料电池应用中铂基催化剂的有前途的替代品。已经通过电子结构方法通过估计来自最强电子元素反应步骤的起始电位来筛选出不同的过渡金属。我们通过第一性原理的微观动力学分析计算了嵌入N-取代石墨烯双空位中的金属原子上的氧还原反应的起始电位。我们发现,对于更多的亲热金属(Cr,Fe,Mn和Ru),纯热力学模型系统地低估了起始电位。此外,亲氧金属(Cr,Fe,Mn和Ru)在反应条件下被氧化,与其还原态相比,导致活性增加。重要的是,共吸附的O m H n物种积极参与反应,这需要对观赏物种进行动态处理。这些发现凸显了电催化过程的热力学分析的局限性,通常对每种金属都假定相同的氧化态,并表明计算电位和实验起始电位之间的偏差不能仅归因于电子结构方法的缺点。


























 京公网安备 11010802027423号
京公网安备 11010802027423号