当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Non-covalent interactions and their impact on the complexation thermodynamics of noble gases with methanol.
Physical Chemistry Chemical Physics ( IF 3.3 ) Pub Date : 2020-07-02 , DOI: 10.1039/d0cp01416h Lúcio Renan Vieira 1 , Sandro Francisco de Brito , Mateus Rodrigues Barbosa , Thiago Oliveira Lopes , Daniel Francisco Scalabrini Machado , Heibbe Cristhian B de Oliveira
Physical Chemistry Chemical Physics ( IF 3.3 ) Pub Date : 2020-07-02 , DOI: 10.1039/d0cp01416h Lúcio Renan Vieira 1 , Sandro Francisco de Brito , Mateus Rodrigues Barbosa , Thiago Oliveira Lopes , Daniel Francisco Scalabrini Machado , Heibbe Cristhian B de Oliveira
Affiliation
|
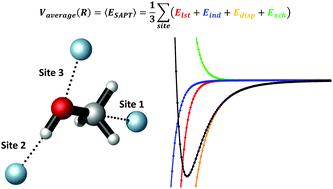
Accurate ab initio calculations provide the reliable information needed to study the potential energy surfaces that control the non-covalent interactions (NCIs) responsible for the formation of weak van der Waals complexes. In this work, relying on the state of the art method for NCI computations, namely symmetry adapted perturbation theory (SAPT), we calculated the potential energy curves for the interaction of noble gases (Ng = He, Ne, Ar and Kr) with methanol in three different interaction sites to account for orientational anisotropy of the interaction potential. Different levels of the SAPT and basis set were employed to disclose the nature of the stabilizing forces acting upon formation of the Ng–CH3OH adducts. SAPT-derived NCIs indicate that dispersion forces are indeed the dominating component of the total energy, but also that induction and electrostatic effects are important to counterbalance the steric repulsions. By solving the Radial Nuclear Schrödinger Equation for the complexes, we also determined the rovibrational structure of the interaction wells to extract invaluable information about the thermodynamic stability of the adducts and how different temperature conditions affect the structure of the dimers. Although SAPT calculations reveal net attractive forces, these do not afford a spontaneous complexation process even at temperatures as low as 40 K.
中文翻译:

非共价相互作用及其对稀有气体与甲醇络合热力学的影响。
准确的从头算起可以提供研究势能面所需的可靠信息,这些势能面控制着构成弱范德华配合物的非共价相互作用(NCI)。在这项工作中,依据用于NCI计算的最新方法,即对称适应扰动理论(SAPT),我们计算了稀有气体(Ng = He,Ne,Ar和Kr)与甲醇相互作用的势能曲线在三个不同的相互作用位点,以解释相互作用势的方向各向异性。采用不同水平的SAPT和基集来揭示作用于Ng-CH 3形成的稳定力的性质OH加合物。SAPT衍生的NCI表明,分散力确实是总能量的主要组成部分,但感应和静电效应对于平衡空间排斥也很重要。通过求解复合物的径向核薛定er方程,我们还确定了相互作用井的振动结构,以提取有关加合物热力学稳定性以及不同温度条件如何影响二聚体结构的宝贵信息。尽管SAPT计算显示出净吸引力,但即使在低至40 K的温度下,这些也无法提供自发的络合过程。
更新日期:2020-08-05
中文翻译:
非共价相互作用及其对稀有气体与甲醇络合热力学的影响。
准确的从头算起可以提供研究势能面所需的可靠信息,这些势能面控制着构成弱范德华配合物的非共价相互作用(NCI)。在这项工作中,依据用于NCI计算的最新方法,即对称适应扰动理论(SAPT),我们计算了稀有气体(Ng = He,Ne,Ar和Kr)与甲醇相互作用的势能曲线在三个不同的相互作用位点,以解释相互作用势的方向各向异性。采用不同水平的SAPT和基集来揭示作用于Ng-CH 3形成的稳定力的性质OH加合物。SAPT衍生的NCI表明,分散力确实是总能量的主要组成部分,但感应和静电效应对于平衡空间排斥也很重要。通过求解复合物的径向核薛定er方程,我们还确定了相互作用井的振动结构,以提取有关加合物热力学稳定性以及不同温度条件如何影响二聚体结构的宝贵信息。尽管SAPT计算显示出净吸引力,但即使在低至40 K的温度下,这些也无法提供自发的络合过程。


























 京公网安备 11010802027423号
京公网安备 11010802027423号