当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
New Molecular-Mechanics Model for Simulations of Hydrogen Fluoride in Chemistry and Biology.
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2020-07-02 , DOI: 10.1021/acs.jctc.0c00247 Esam A Orabi 1 , José D Faraldo-Gómez 1
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2020-07-02 , DOI: 10.1021/acs.jctc.0c00247 Esam A Orabi 1 , José D Faraldo-Gómez 1
Affiliation
|
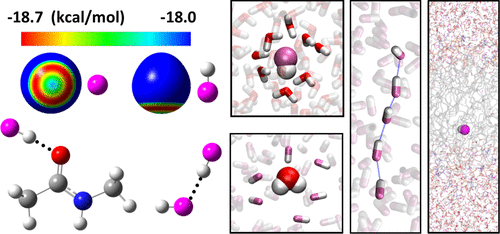
Hydrogen fluoride (HF) is the most polar diatomic molecule and one of the simplest molecules capable of hydrogen-bonding. HF deviates from ideality both in the gas phase and in solution and is thus of great interest from a fundamental standpoint. Pure and aqueous HF solutions are broadly used in chemical and industrial processes, despite their high toxicity. HF is a stable species also in some biological conditions, because it does not readily dissociate in water unlike other hydrogen halides; yet, little is known about how HF interacts with biomolecules. Here, we set out to develop a molecular-mechanics model to enable computer simulations of HF in chemical and biological applications. This model is based on a comprehensive high-level ab initio quantum chemical investigation of the structure and energetics of the HF monomer and dimer; (HF)n clusters, for n = 3–7; various clusters of HF and H2O; and complexes of HF with analogs of all 20 amino acids and of several commonly occurring lipids, both neutral and ionized. This systematic analysis explains the unique properties of this molecule: for example, that interacting HF molecules favor nonlinear geometries despite being diatomic and that HF is a strong H-bond donor but a poor acceptor. The ab initio data also enables us to calibrate a three-site molecular-mechanics model, with which we investigate the structure and thermodynamic properties of gaseous, liquid, and supercritical HF in a wide range of temperatures and pressures; the solvation structure of HF in water and of H2O in liquid HF; and the free diffusion of HF across a lipid bilayer, a key process underlying the high cytotoxicity of HF. Despite its inherent simplifications, the model presented significantly improves upon previous efforts to capture the properties of pure and aqueous HF fluids by molecular-mechanics methods and to our knowledge constitutes the first parameter set calibrated for biomolecular simulations.
中文翻译:

用于在化学和生物学中模拟氟化氢的新分子力学模型。
氟化氢 (HF) 是极性最强的双原子分子,也是最简单的能够形成氢键的分子之一。HF 在气相和溶液中都偏离了理想状态,因此从基本的角度来看是非常有趣的。纯的和水性 HF 溶液广泛用于化学和工业过程,尽管它们具有高毒性。HF 在某些生物条件下也是一种稳定的物质,因为与其他卤化氢不同,它在水中不易解离;然而,人们对 HF 如何与生物分子相互作用知之甚少。在这里,我们着手开发一个分子力学模型,以便在化学和生物应用中对 HF 进行计算机模拟。该模型基于对 HF 单体和二聚体的结构和能量学的全面高级 ab initio 量子化学研究;(高频)n 个簇,对于n = 3–7;各种HF和H 2 O簇;HF 与所有 20 种氨基酸和几种常见脂质的类似物(中性和离子化)的复合物。这种系统分析解释了该分子的独特性质:例如,尽管是双原子的,但相互作用的 HF 分子有利于非线性几何形状,并且 HF 是强氢键供体但不是受体。ab initio 数据还使我们能够校准三点分子力学模型,通过该模型我们可以研究气体、液体和超临界 HF 在广泛的温度和压力范围内的结构和热力学特性;HF在水中和H 2的溶剂化结构液体HF中的O;以及 HF 在脂双层中的自由扩散,这是 HF 高细胞毒性的关键过程。尽管存在固有的简化,但该模型显着改进了以前通过分子力学方法捕获纯和水性 HF 流体特性的努力,并且据我们所知,它构成了第一个为生物分子模拟校准的参数集。
更新日期:2020-08-11
中文翻译:
用于在化学和生物学中模拟氟化氢的新分子力学模型。
氟化氢 (HF) 是极性最强的双原子分子,也是最简单的能够形成氢键的分子之一。HF 在气相和溶液中都偏离了理想状态,因此从基本的角度来看是非常有趣的。纯的和水性 HF 溶液广泛用于化学和工业过程,尽管它们具有高毒性。HF 在某些生物条件下也是一种稳定的物质,因为与其他卤化氢不同,它在水中不易解离;然而,人们对 HF 如何与生物分子相互作用知之甚少。在这里,我们着手开发一个分子力学模型,以便在化学和生物应用中对 HF 进行计算机模拟。该模型基于对 HF 单体和二聚体的结构和能量学的全面高级 ab initio 量子化学研究;(高频)n 个簇,对于n = 3–7;各种HF和H 2 O簇;HF 与所有 20 种氨基酸和几种常见脂质的类似物(中性和离子化)的复合物。这种系统分析解释了该分子的独特性质:例如,尽管是双原子的,但相互作用的 HF 分子有利于非线性几何形状,并且 HF 是强氢键供体但不是受体。ab initio 数据还使我们能够校准三点分子力学模型,通过该模型我们可以研究气体、液体和超临界 HF 在广泛的温度和压力范围内的结构和热力学特性;HF在水中和H 2的溶剂化结构液体HF中的O;以及 HF 在脂双层中的自由扩散,这是 HF 高细胞毒性的关键过程。尽管存在固有的简化,但该模型显着改进了以前通过分子力学方法捕获纯和水性 HF 流体特性的努力,并且据我们所知,它构成了第一个为生物分子模拟校准的参数集。

























 京公网安备 11010802027423号
京公网安备 11010802027423号