当前位置:
X-MOL 学术
›
Chem. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Large transition state stabilization from a weak hydrogen bond
Chemical Science ( IF 8.4 ) Pub Date : 2020-07-02 , DOI: 10.1039/d0sc02806a Erik C. Vik 1, 2, 3, 4 , Ping Li 1, 2, 3, 4 , Josef M. Maier 1, 2, 3, 4 , Daniel O. Madukwe 1, 2, 3, 4 , Vitaly A. Rassolov 1, 2, 3, 4 , Perry J. Pellechia 1, 2, 3, 4 , Eric Masson 1, 4, 5, 6 , Ken D. Shimizu 1, 2, 3, 4
Chemical Science ( IF 8.4 ) Pub Date : 2020-07-02 , DOI: 10.1039/d0sc02806a Erik C. Vik 1, 2, 3, 4 , Ping Li 1, 2, 3, 4 , Josef M. Maier 1, 2, 3, 4 , Daniel O. Madukwe 1, 2, 3, 4 , Vitaly A. Rassolov 1, 2, 3, 4 , Perry J. Pellechia 1, 2, 3, 4 , Eric Masson 1, 4, 5, 6 , Ken D. Shimizu 1, 2, 3, 4
Affiliation
|
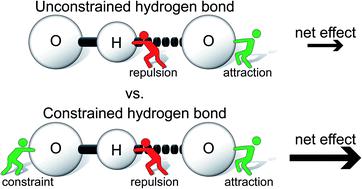
A series of molecular rotors was designed to study and measure the rate accelerating effects of an intramolecular hydrogen bond. The rotors form a weak neutral O–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bond in the planar transition state (TS) of the bond rotation process. The rotational barrier of the hydrogen bonding rotors was dramatically lower (9.9 kcal mol−1) than control rotors which could not form hydrogen bonds. The magnitude of the stabilization was significantly larger than predicted based on the independently measured strength of a similar O–H⋯OC hydrogen bond (1.5 kcal mol−1). The origins of the large transition state stabilization were studied via experimental substituent effect and computational perturbation analyses. Energy decomposition analysis of the hydrogen bonding interaction revealed a significant reduction in the repulsive component of the hydrogen bonding interaction. The rigid framework of the molecular rotors positions and preorganizes the interacting groups in the transition state. This study demonstrates that with proper design a single hydrogen bond can lead to a TS stabilization that is greater than the intrinsic interaction energy, which has applications in catalyst design and in the study of enzyme mechanisms.
C hydrogen bond in the planar transition state (TS) of the bond rotation process. The rotational barrier of the hydrogen bonding rotors was dramatically lower (9.9 kcal mol−1) than control rotors which could not form hydrogen bonds. The magnitude of the stabilization was significantly larger than predicted based on the independently measured strength of a similar O–H⋯OC hydrogen bond (1.5 kcal mol−1). The origins of the large transition state stabilization were studied via experimental substituent effect and computational perturbation analyses. Energy decomposition analysis of the hydrogen bonding interaction revealed a significant reduction in the repulsive component of the hydrogen bonding interaction. The rigid framework of the molecular rotors positions and preorganizes the interacting groups in the transition state. This study demonstrates that with proper design a single hydrogen bond can lead to a TS stabilization that is greater than the intrinsic interaction energy, which has applications in catalyst design and in the study of enzyme mechanisms.
中文翻译:

弱氢键可实现较大的过渡态稳定性
设计了一系列分子转子,以研究和测量分子内氢键的加速作用。转子在键旋转过程的平面过渡状态(TS)中形成弱的中性O–H⋯O C氢键。氢键转子的旋转势垒比不能形成氢键的控制转子低得多(9.9 kcal mol -1)。稳定的幅度明显大于根据类似的O–H⋯O C氢键(1.5 kcal mol -1)的独立测量强度所预测的值。大过渡态稳定的起源是通过实验性取代基效应和计算扰动分析。氢键相互作用的能量分解分析表明,氢键相互作用的斥力成分显着降低。分子转子的刚性框架在过渡态中定位并预组织了相互作用的基团。这项研究表明,通过适当的设计,单个氢键可以导致大于固有相互作用能的TS稳定化,这在催化剂设计和酶机理研究中具有应用。
更新日期:2020-07-22
C hydrogen bond in the planar transition state (TS) of the bond rotation process. The rotational barrier of the hydrogen bonding rotors was dramatically lower (9.9 kcal mol−1) than control rotors which could not form hydrogen bonds. The magnitude of the stabilization was significantly larger than predicted based on the independently measured strength of a similar O–H⋯OC hydrogen bond (1.5 kcal mol−1). The origins of the large transition state stabilization were studied via experimental substituent effect and computational perturbation analyses. Energy decomposition analysis of the hydrogen bonding interaction revealed a significant reduction in the repulsive component of the hydrogen bonding interaction. The rigid framework of the molecular rotors positions and preorganizes the interacting groups in the transition state. This study demonstrates that with proper design a single hydrogen bond can lead to a TS stabilization that is greater than the intrinsic interaction energy, which has applications in catalyst design and in the study of enzyme mechanisms.
中文翻译:
弱氢键可实现较大的过渡态稳定性
设计了一系列分子转子,以研究和测量分子内氢键的加速作用。转子
在键旋转过程的平面过渡状态(TS)中形成弱的中性O–H⋯O C氢键。氢键转子的旋转势垒比不能形成氢键的控制转子低得多(9.9 kcal mol -1)。稳定的幅度明显大于根据类似的O–H⋯O C氢键(1.5 kcal mol -1)的独立测量强度所预测的值。大过渡态稳定的起源是通过实验性取代基效应和计算扰动分析。氢键相互作用的能量分解分析表明,氢键相互作用的斥力成分显着降低。分子转子的刚性框架在过渡态中定位并预组织了相互作用的基团。这项研究表明,通过适当的设计,单个氢键可以导致大于固有相互作用能的TS稳定化,这在催化剂设计和酶机理研究中具有应用。

























 京公网安备 11010802027423号
京公网安备 11010802027423号