当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Modified Lennard‐Jones potentials for nanoscale atoms
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2020-06-27 , DOI: 10.1002/jcc.26368 Celina Sikorska 1 , Nicola Gaston 1
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2020-06-27 , DOI: 10.1002/jcc.26368 Celina Sikorska 1 , Nicola Gaston 1
Affiliation
|
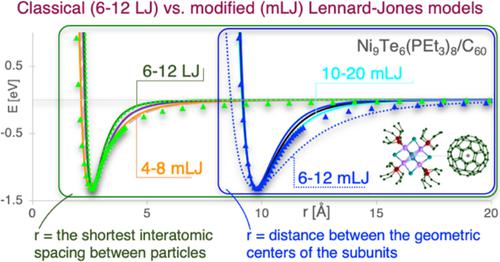
A classical 6–12 Lennard‐Jones (LJ) equation has been widely used to model materials and is the potential of choice in studies when the focus is on fundamental issues. Here we report a systematic study comparing the pair interaction potentials within solid‐state materials (i.e., [Co6Se8(PEt3)6][C60]2, [Cr6Te8(PEt3)6][C60]2, [Ni9Te6(PEt3)8][C60]) using density functional theory (DFT) calculations and LJ parametrization. Both classical (6–12 LJ) and modified LJ (mLJ) models were developed. In the mLJ approach, the exponents 6 and 12 are replaced by different integer number n and 2n, respectively, and an additional parameter (α) is introduced to describe intermolecular distance shift arising within the geometric centers' approach (instead of the shortest interatomic distance between particles). A general LJ approach reexamination reveals that in the case of nanoatoms, the attractive term decays with distance as the inverse fourth power, and the dominating at short distances repulsive term decays as the inverse eighth power. The modification of the LJ equation is even more prominent for interaction profiles, where intermolecular distance corresponds to separation between geometric centers of particles. In this approach, the attractive term decays with distance as the inverse 12th power, while the repulsive term decays rapidly (as the inverse 24th power). Thus, the mLJ models (e.g., 4–8 LJ) rather than the 6–12 classical ones seem to be a better choice for the description of binary interactions of nanoatoms. The developed mLJ models and electronic structure characteristics give an insight into the explanation of the unique physicochemical properties of superatomic‐based solid‐state materials.
中文翻译:

纳米级原子的修正 Lennard-Jones 势
经典的 6-12 Lennard-Jones (LJ) 方程已广泛用于材料建模,并且是研究重点关注基本问题时的潜在选择。在这里,我们报告了一项比较固态材料(即 [Co6Se8(PEt3)6][C60]2、[Cr6Te8(PEt3)6][C60]2、[Ni9Te6(PEt3)8] 中的对相互作用势的系统研究) [C60]) 使用密度泛函理论 (DFT) 计算和 LJ 参数化。开发了经典 (6-12 LJ) 和改进的 LJ (mLJ) 模型。在 mLJ 方法中,指数 6 和 12 分别由不同的整数 n 和 2n 代替,并引入了一个附加参数 (α) 来描述几何中心方法内产生的分子间距离偏移(而不是最短原子间距离)粒子之间)。一般的 LJ 方法重新检查表明,在纳米原子的情况下,吸引力项随距离衰减为倒数四次方,而在短距离占主导地位的排斥项衰减为倒数八次方。LJ 方程的修改对于相互作用曲线更为突出,其中分子间距离对应于粒子几何中心之间的间隔。在这种方法中,吸引力项随距离衰减的 12 次方,而排斥项迅速衰减(24 次方)。因此,mLJ 模型(例如,4-8 LJ)而不是 6-12 经典模型似乎是描述纳米原子二元相互作用的更好选择。
更新日期:2020-06-27
中文翻译:
纳米级原子的修正 Lennard-Jones 势
经典的 6-12 Lennard-Jones (LJ) 方程已广泛用于材料建模,并且是研究重点关注基本问题时的潜在选择。在这里,我们报告了一项比较固态材料(即 [Co6Se8(PEt3)6][C60]2、[Cr6Te8(PEt3)6][C60]2、[Ni9Te6(PEt3)8] 中的对相互作用势的系统研究) [C60]) 使用密度泛函理论 (DFT) 计算和 LJ 参数化。开发了经典 (6-12 LJ) 和改进的 LJ (mLJ) 模型。在 mLJ 方法中,指数 6 和 12 分别由不同的整数 n 和 2n 代替,并引入了一个附加参数 (α) 来描述几何中心方法内产生的分子间距离偏移(而不是最短原子间距离)粒子之间)。一般的 LJ 方法重新检查表明,在纳米原子的情况下,吸引力项随距离衰减为倒数四次方,而在短距离占主导地位的排斥项衰减为倒数八次方。LJ 方程的修改对于相互作用曲线更为突出,其中分子间距离对应于粒子几何中心之间的间隔。在这种方法中,吸引力项随距离衰减的 12 次方,而排斥项迅速衰减(24 次方)。因此,mLJ 模型(例如,4-8 LJ)而不是 6-12 经典模型似乎是描述纳米原子二元相互作用的更好选择。

























 京公网安备 11010802027423号
京公网安备 11010802027423号