当前位置:
X-MOL 学术
›
Acta Cryst. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Crystal and geometry-optimized structure of an anthracene-based Diels-Alder adduct.
Acta Crystallographica Section C ( IF 0.8 ) Pub Date : 2020-06-26 , DOI: 10.1107/s2053229620008128 Zachary E Hillman 1 , Joseph M Tanski 2 , Andrea Roberts 1
Acta Crystallographica Section C ( IF 0.8 ) Pub Date : 2020-06-26 , DOI: 10.1107/s2053229620008128 Zachary E Hillman 1 , Joseph M Tanski 2 , Andrea Roberts 1
Affiliation
|
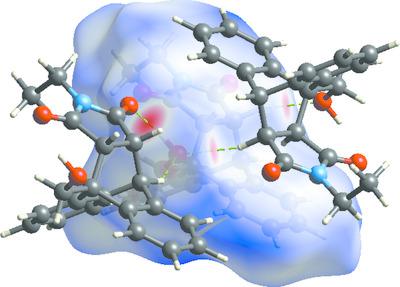
Computational calculations of an anthracene‐based Diels–Alder adduct, namely, 17‐ethyl‐1‐hydroxymethyl‐17‐azapentacyclo[6.6.5.02,7.09,14.015,19]nonadeca‐2,4,6,9,11,13‐hexaene‐16,18‐dione, C21H19NO3, predicting density functional theory (DFT) optimized geometries in the gas phase are compared in terms of accuracy relative to the solid‐state crystal structure and computational cost. Crystal structure determination and Hirshfeld surface analysis of the racemic product reveal that the molecules are linked by O—H…O=C hydrogen bonds between the hydroxy and carbonyl groups, accounting for 9.5% of the intermolecular contacts, while H…H contacts represent 56.9% of the total. Boltzmann population analysis of computed relative rotamer abundances in the gas phase are based on lower‐level geometry optimization and thermochemical corrections coupled with higher‐level electronic energy calculations using the B2PLYP double‐hybrid functional. As expected, the choice of density functional has a greater effect than the basis set on accuracy for all levels of theory. For any given functional, increasing the basis set size did not always correlate with increasingly accurate structures. The hybrid functional B3LYP without dispersion correction routinely gave the closest approximations to the crystal structure where the B3LYP/aug‐cc‐pVDZ combination afforded the best structure (r.m.s. deviation = 0.1314 Å). However, the B3LYP/6‐31+G(d,p) level of theory represents the best compromise between accuracy (r.m.s. deviation = 0.1388 Å) and cost as it yielded appreciably accurate results in a fraction of the time compared to other method combinations.
中文翻译:

蒽基Diels-Alder加合物的晶体和几何优化结构。
蒽基Diels-Alder加合物的计算计算,即17-乙基-1-羟甲基-17-氮杂五环[6.6.5.0 2,7 .0 9,14 .0 15,19 ] nonadeca-2,4,6 ,9,11,13-己烯-16,18-二酮,C 21 H 19 NO 3,在相对于固态晶体结构的准确性和计算成本方面,比较了预测密度泛函理论(DFT)在气相中优化的几何形状。外消旋产物的晶体结构测定和Hirshfeld表面分析表明,这些分子通过羟基与羰基之间的OH-H…O = C氢键连接,占分子间接触的9.5%,而H…H接触占56.9。占总数的百分比。气相中相对旋转异构体丰度的玻尔兹曼种群分析是基于较低级的几何优化和热化学校正,以及使用B2PLYP双混合函数进行的较高级电子能量计算。不出所料 在所有理论水平上,密度泛函的选择都比基于精度设定的影响更大。对于任何给定的功能,增加基础集的大小并不总是与日益精确的结构相关。通常不进行色散校正的混合功能B3LYP可以最接近晶体结构,其中B3LYP / aug-cc-pVDZ组合提供了最佳结构(均方根偏差= 0.1314Å)。但是,B3LYP / 6‐31 + G(d,p)的理论水平代表了准确度(均方根偏差= 0.1388Å)和成本之间的最佳折衷,因为与其他方法组合相比,它可以在很短的时间内产生相当准确的结果。常规的没有色散校正的混合功能B3LYP通常提供最接近晶体结构的近似结构,其中B3LYP / aug-cc-pVDZ组合提供了最佳结构(均方根偏差= 0.1314Å)。但是,B3LYP / 6‐31 + G(d,p)的理论水平代表了精度(均方根偏差= 0.1388Å)和成本之间的最佳折衷,因为与其他方法组合相比,它在短短的时间内就可以产生相当准确的结果。通常不进行色散校正的混合功能B3LYP可以最接近晶体结构,其中B3LYP / aug-cc-pVDZ组合提供了最佳结构(均方根偏差= 0.1314Å)。但是,B3LYP / 6‐31 + G(d,p)的理论水平代表了精度(均方根偏差= 0.1388Å)和成本之间的最佳折衷,因为与其他方法组合相比,它在短短的时间内就可以产生相当准确的结果。
更新日期:2020-06-26
中文翻译:
蒽基Diels-Alder加合物的晶体和几何优化结构。
蒽基Diels-Alder加合物的计算计算,即17-乙基-1-羟甲基-17-氮杂五环[6.6.5.0 2,7 .0 9,14 .0 15,19 ] nonadeca-2,4,6 ,9,11,13-己烯-16,18-二酮,C 21 H 19 NO 3,在相对于固态晶体结构的准确性和计算成本方面,比较了预测密度泛函理论(DFT)在气相中优化的几何形状。外消旋产物的晶体结构测定和Hirshfeld表面分析表明,这些分子通过羟基与羰基之间的OH-H…O = C氢键连接,占分子间接触的9.5%,而H…H接触占56.9。占总数的百分比。气相中相对旋转异构体丰度的玻尔兹曼种群分析是基于较低级的几何优化和热化学校正,以及使用B2PLYP双混合函数进行的较高级电子能量计算。不出所料 在所有理论水平上,密度泛函的选择都比基于精度设定的影响更大。对于任何给定的功能,增加基础集的大小并不总是与日益精确的结构相关。通常不进行色散校正的混合功能B3LYP可以最接近晶体结构,其中B3LYP / aug-cc-pVDZ组合提供了最佳结构(均方根偏差= 0.1314Å)。但是,B3LYP / 6‐31 + G(d,p)的理论水平代表了准确度(均方根偏差= 0.1388Å)和成本之间的最佳折衷,因为与其他方法组合相比,它可以在很短的时间内产生相当准确的结果。常规的没有色散校正的混合功能B3LYP通常提供最接近晶体结构的近似结构,其中B3LYP / aug-cc-pVDZ组合提供了最佳结构(均方根偏差= 0.1314Å)。但是,B3LYP / 6‐31 + G(d,p)的理论水平代表了精度(均方根偏差= 0.1388Å)和成本之间的最佳折衷,因为与其他方法组合相比,它在短短的时间内就可以产生相当准确的结果。通常不进行色散校正的混合功能B3LYP可以最接近晶体结构,其中B3LYP / aug-cc-pVDZ组合提供了最佳结构(均方根偏差= 0.1314Å)。但是,B3LYP / 6‐31 + G(d,p)的理论水平代表了精度(均方根偏差= 0.1388Å)和成本之间的最佳折衷,因为与其他方法组合相比,它在短短的时间内就可以产生相当准确的结果。


























 京公网安备 11010802027423号
京公网安备 11010802027423号