Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Chemical vapour deposition of graphene on copper-nickel alloys: the simulation of a thermodynamic and kinetic approach.
Nanoscale ( IF 6.7 ) Pub Date : 2020-06-23 , DOI: 10.1039/d0nr00302f Samir H Al-Hilfi 1 , Brian Derby , Philip A Martin , J Christopher Whitehead
Nanoscale ( IF 6.7 ) Pub Date : 2020-06-23 , DOI: 10.1039/d0nr00302f Samir H Al-Hilfi 1 , Brian Derby , Philip A Martin , J Christopher Whitehead
Affiliation
|
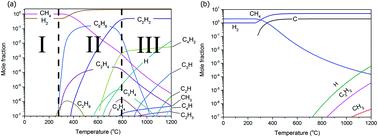
Chemical vapour deposition (CVD) of graphene on transition metals is generally believed to be the fabrication route best suited for the production of high-quality large-area graphene sheets. The mechanism of CVD graphene growth is governed by interactions in both the gas phase and at the surface. Here we present a simulation of the CVD graphene growth mechanism which includes thermodynamics, gas phase kinetics and the surface reaction in a sequential manner. The thermodynamic simulation shows that the deposition driving force is the greatest for high carbon to hydrogen ratios and reaches a maximum at around 850 °C. No graphene growth is observed below this temperature. The surface kinetic model also shows that below this temperature, the carbon surface concentration is less than the solubility limit, thus no film can grow. The effect of the reaction chamber geometry on the product concentrations was clear from the gas phase decomposition reactions. The gas residence times studied here (around 0.07 s) show that the optimum gas phase composition is far from that expected at thermodynamic equilibrium. The surface kinetics of CH4 reactions on Ni, Cu and Cu–Ni surfaces shows good agreement with the experimental results for different growth pressures (0.1 to 0.7 mbar), temperatures (600 to 1200 °C) and different Ni thicknesses (25–500 μm). Also, the model works well when substrates with various C solubilities are used. The thermodynamic and kinetic models described here can be used for the design of improved reactors to optimise the production of graphene with differing qualities, either single or multi-layer and sizes. More importantly, the transfer to a continuous process with a moving substrate should also be possible using the model if it is extended from 2D to 3D.
中文翻译:

石墨烯在铜-镍合金上的化学气相沉积:热力学和动力学方法的模拟。
石墨烯在过渡金属上的化学气相沉积(CVD)通常被认为是最适合于生产高质量大面积石墨烯片的制造途径。CVD石墨烯生长的机理受气相和表面相互作用的支配。在这里,我们介绍了CVD石墨烯生长机理的模拟,包括依次进行的热力学,气相动力学和表面反应。热力学模拟表明,沉积驱动力对于高碳氢比最大,并在850°C左右达到最大值。在该温度以下没有观察到石墨烯生长。表面动力学模型还表明,在此温度以下,碳表面浓度小于溶解度极限,因此没有膜可以生长。从气相分解反应可以明显看出反应室几何形状对产物浓度的影响。在此研究的气体停留时间(约0.07 s)表明,最佳气相组成与热力学平衡时的预期相差甚远。CH的表面动力学在不同的生长压力(0.1至0.7 mbar),温度(600至1200°C)和不同的镍厚度(25-500μm)下,Ni,Cu和Cu-Ni表面的4种反应与实验结果显示出良好的一致性。同样,当使用具有各种C溶解度的底物时,该模型也很好用。此处描述的热力学和动力学模型可用于设计改进的反应器,以优化具有不同质量(单层或多层和尺寸)的石墨烯的生产。更重要的是,如果模型是从2D扩展到3D,则还可以使用该模型将其转移到具有移动基板的连续过程中。
更新日期:2020-07-23
中文翻译:
石墨烯在铜-镍合金上的化学气相沉积:热力学和动力学方法的模拟。
石墨烯在过渡金属上的化学气相沉积(CVD)通常被认为是最适合于生产高质量大面积石墨烯片的制造途径。CVD石墨烯生长的机理受气相和表面相互作用的支配。在这里,我们介绍了CVD石墨烯生长机理的模拟,包括依次进行的热力学,气相动力学和表面反应。热力学模拟表明,沉积驱动力对于高碳氢比最大,并在850°C左右达到最大值。在该温度以下没有观察到石墨烯生长。表面动力学模型还表明,在此温度以下,碳表面浓度小于溶解度极限,因此没有膜可以生长。从气相分解反应可以明显看出反应室几何形状对产物浓度的影响。在此研究的气体停留时间(约0.07 s)表明,最佳气相组成与热力学平衡时的预期相差甚远。CH的表面动力学在不同的生长压力(0.1至0.7 mbar),温度(600至1200°C)和不同的镍厚度(25-500μm)下,Ni,Cu和Cu-Ni表面的4种反应与实验结果显示出良好的一致性。同样,当使用具有各种C溶解度的底物时,该模型也很好用。此处描述的热力学和动力学模型可用于设计改进的反应器,以优化具有不同质量(单层或多层和尺寸)的石墨烯的生产。更重要的是,如果模型是从2D扩展到3D,则还可以使用该模型将其转移到具有移动基板的连续过程中。


























 京公网安备 11010802027423号
京公网安备 11010802027423号