当前位置:
X-MOL 学术
›
Cryst. Growth Des.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
The C–I···–O–N+ Halogen Bonds with Tetraiodoethylene and Aromatic N-Oxides
Crystal Growth & Design ( IF 3.8 ) Pub Date : 2020-06-18 , DOI: 10.1021/acs.cgd.0c00560 Khai-Nghi Truong 1 , J. Mikko Rautiainen 1 , Kari Rissanen 1 , Rakesh Puttreddy 1, 2
Crystal Growth & Design ( IF 3.8 ) Pub Date : 2020-06-18 , DOI: 10.1021/acs.cgd.0c00560 Khai-Nghi Truong 1 , J. Mikko Rautiainen 1 , Kari Rissanen 1 , Rakesh Puttreddy 1, 2
Affiliation
|
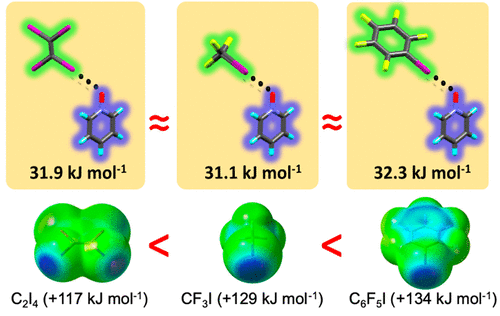
The nature of C–I···–O–N+ interactions, the first of its kind, between nonfluorinated tetraiodoethylene halogen bond (XB) donor and pyridine N-oxides (PyNO) are studied by single-crystal X-ray diffraction and density functional theory (DFT) calculations. Despite the nonfluorinated nature of the C2I4, the I···O halogen bond distances are similar to well-known perfluorohaloalkane/-arene donor-PyNO analogues. With C2I4, oxygens of the N-oxides adopt exclusively μ2-XB coordination in contrast to the versatile bonding modes observed with perfluorinated XB donors. The C2I4 as the XB donor forms with PyNO’s one-dimensional chain polymer structures in which the C2I4···(μ-PyNO)2···C2I4 segments manifest two bonding motifs, namely, side-by-side (vicinal di-iodo) and head-to-head (geminal di-iodo), due to the nearly symmetric square-planar structure of the C2I4. While the attractive nature between I and O atoms is mainly electrostatic, the narrow range of C···O bond parameters demonstrates that the π-bond between four iodine atoms also plays an important role in enhancing the σ-hole strength. DFT-Based monodentate XB interaction energies, ΔEint, in 13 1:1 XB complexes vary between 31.9 and 46.5 kJ mol–1, the strongest remarkably exceeding the value reported for I–I···–O–N+ = 42.0 kJ mol–1. In the case of C2I4·(pyridine N-oxide) [31.9 kJ mol–1], the monodentate XB energy is on a par with perfluorinated donor complexes, namely, CF3I·(pyridine N-oxide) [31.1 kJ mol–1] and C6F5I·(pyridine N-oxide) [32.3 kJ mol–1].
中文翻译:

C–I··· – O–N +与四碘乙烯和芳香族N-氧化物的卤素键
的性质C-1 ... - O-N +相互作用,首先它的种,非氟化四碘卤键(XB)供体和吡啶N-氧化物(PyNO)之间通过单晶X射线衍射进行研究,并密度泛函理论(DFT)计算。尽管C 2 I 4具有非氟化性质,但I··O卤素键的距离与众所周知的全氟卤代烷/芳烃供体-PyNO类似物相似。用C 2我4,N-氧化物的氧采用专门μ 2与用全氟化XB供体中观察到的多功能键方式-XB协调。C 2我4因为XB供体具有PyNO的一维链状聚合物结构,其中C 2 I 4 ···(μ- PyNO)2 ···C 2 I 4片段表现出两个键合基序,即并排(由于C 2 I 4几乎是对称的正方形平面结构,因此,“邻二碘”和“头对头”(二碘)。虽然I和O原子之间的吸引性质主要是静电,但C···O键参数的范围狭窄,这表明四个碘原子之间的π键在增强σ空穴强度方面也起着重要作用。基于DFT的单齿XB相互作用能ΔE int在13个1:1 XB络合物中的变化范围为31.9至46.5 kJ mol –1,最强的超过了I–I·· – O–N + = 42.0 kJ mol –1的报告值。在C 2 I 4 ·(吡啶N-氧化物)[31.9 kJ mol –1 ]的情况下,单齿XB能量与全氟化供体配合物CF 3 I·(吡啶N-氧化物)[31.1 ]相当。kJ mol –1 ]和C 6 F 5 I·(吡啶N-氧化物)[32.3 kJ mol –1 ]。
更新日期:2020-08-05
中文翻译:
C–I··· – O–N +与四碘乙烯和芳香族N-氧化物的卤素键
的性质C-1 ... - O-N +相互作用,首先它的种,非氟化四碘卤键(XB)供体和吡啶N-氧化物(PyNO)之间通过单晶X射线衍射进行研究,并密度泛函理论(DFT)计算。尽管C 2 I 4具有非氟化性质,但I··O卤素键的距离与众所周知的全氟卤代烷/芳烃供体-PyNO类似物相似。用C 2我4,N-氧化物的氧采用专门μ 2与用全氟化XB供体中观察到的多功能键方式-XB协调。C 2我4因为XB供体具有PyNO的一维链状聚合物结构,其中C 2 I 4 ···(μ- PyNO)2 ···C 2 I 4片段表现出两个键合基序,即并排(由于C 2 I 4几乎是对称的正方形平面结构,因此,“邻二碘”和“头对头”(二碘)。虽然I和O原子之间的吸引性质主要是静电,但C···O键参数的范围狭窄,这表明四个碘原子之间的π键在增强σ空穴强度方面也起着重要作用。基于DFT的单齿XB相互作用能ΔE int在13个1:1 XB络合物中的变化范围为31.9至46.5 kJ mol –1,最强的超过了I–I·· – O–N + = 42.0 kJ mol –1的报告值。在C 2 I 4 ·(吡啶N-氧化物)[31.9 kJ mol –1 ]的情况下,单齿XB能量与全氟化供体配合物CF 3 I·(吡啶N-氧化物)[31.1 ]相当。kJ mol –1 ]和C 6 F 5 I·(吡啶N-氧化物)[32.3 kJ mol –1 ]。

























 京公网安备 11010802027423号
京公网安备 11010802027423号