当前位置:
X-MOL 学术
›
Chem. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Deciphering the origin of million-fold reactivity observed for the open core diiron [HO–FeIII–O–FeIVO]2+ species towards C–H bond activation: role of spin-states, spin-coupling, and spin-cooperation
Chemical Science ( IF 8.4 ) Pub Date : 2020-06-18 , DOI: 10.1039/d0sc02624g Mursaleem Ansari 1 , Dhurairajan Senthilnathan 2 , Gopalan Rajaraman 1
Chemical Science ( IF 8.4 ) Pub Date : 2020-06-18 , DOI: 10.1039/d0sc02624g Mursaleem Ansari 1 , Dhurairajan Senthilnathan 2 , Gopalan Rajaraman 1
Affiliation
|
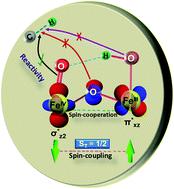
High-valent metal–oxo species have been characterised as key intermediates in both heme and non-heme enzymes that are found to perform efficient aliphatic hydroxylation, epoxidation, halogenation, and dehydrogenation reactions. Several biomimetic model complexes have been synthesised over the years to mimic both the structure and function of metalloenzymes. The diamond-core [Fe2(μ-O)2] is one of the celebrated models in this context as this has been proposed as the catalytically active species in soluble methane monooxygenase enzymes (sMMO), which perform the challenging chemical conversion of methane to methanol at ease. In this context, a report of open core [HO(L)FeIII–O–FeIV(O)(L)]2+ (1) gains attention as this activates C–H bonds a million-fold faster compared to the diamond-core structure and has the dual catalytic ability to perform hydroxylation as well as desaturation with organic substrates. In this study, we have employed density functional methods to probe the origin of the very high reactivity observed for this complex and also to shed light on how this complex performs efficient hydroxylation and desaturation of alkanes. By modelling fifteen possible spin-states for 1 that could potentially participate in the reaction mechanism, our calculations reveal a doublet ground state for 1 arising from antiferromagnetic coupling between the quartet FeIV centre and the sextet FeIII centre, which regulates the reactivity of this species. The unusual stabilisation of the high-spin ground state for FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O is due to the strong overlap of
O is due to the strong overlap of 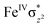 with the
with the 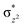 orbital, reducing the antibonding interactions via spin-cooperation. The electronic structure features computed for 1 are consistent with experiments offering confidence in the methodology chosen. Further, we have probed various mechanistic pathways for the C–H bond activation as well as –OH rebound/desaturation of alkanes. An extremely small barrier height computed for the first hydrogen atom abstraction by the terminal FeIVO unit was found to be responsible for the million-fold activation observed in the experiments. The barrier height computed for –OH rebound by the FeIII–OH unit is also smaller suggesting a facile hydroxylation of organic substrates by 1. A strong spin-cooperation between the two iron centres also reduces the barrier for second hydrogen atom abstraction, thus making the desaturation pathway competitive. Both the spin-state as well as spin-coupling between the two metal centres play a crucial role in dictating the reactivity for species 1. By exploring various mechanistic pathways, our study unveils the fact that the bridged μ-oxo group is a poor electrophile for both C–H activation as well for –OH rebound. As more and more evidence is gathered in recent years for the open core geometry of sMMO enzymes, the idea of enhancing the reactivity via an open-core motif has far-reaching consequences.
orbital, reducing the antibonding interactions via spin-cooperation. The electronic structure features computed for 1 are consistent with experiments offering confidence in the methodology chosen. Further, we have probed various mechanistic pathways for the C–H bond activation as well as –OH rebound/desaturation of alkanes. An extremely small barrier height computed for the first hydrogen atom abstraction by the terminal FeIVO unit was found to be responsible for the million-fold activation observed in the experiments. The barrier height computed for –OH rebound by the FeIII–OH unit is also smaller suggesting a facile hydroxylation of organic substrates by 1. A strong spin-cooperation between the two iron centres also reduces the barrier for second hydrogen atom abstraction, thus making the desaturation pathway competitive. Both the spin-state as well as spin-coupling between the two metal centres play a crucial role in dictating the reactivity for species 1. By exploring various mechanistic pathways, our study unveils the fact that the bridged μ-oxo group is a poor electrophile for both C–H activation as well for –OH rebound. As more and more evidence is gathered in recent years for the open core geometry of sMMO enzymes, the idea of enhancing the reactivity via an open-core motif has far-reaching consequences.
中文翻译:

破译开核二铁 [HO-FeIII-O-FeIVO]2+ 物种对 C-H 键活化观察到的百万倍反应性的起源:自旋态、自旋耦合和自旋合作的作用
高价金属-氧代物种已被表征为血红素和非血红素酶的关键中间体,被发现可进行有效的脂肪族羟基化、环氧化、卤化和脱氢反应。多年来已经合成了几种仿生模型复合物来模拟金属酶的结构和功能。金刚石核心 [Fe 2 (μ-O) 2 ] 是这方面著名的模型之一,因为它已被提议作为可溶性甲烷单加氧酶 (sMMO) 中的催化活性物质,它执行甲烷的具有挑战性的化学转化对甲醇放心。在此背景下,关于开核 [HO(L)Fe III –O–Fe IV (O)(L)] 2+ ( 1) 受到关注,因为与金刚石核心结构相比,它激活 C-H 键的速度快了一百万倍,并且具有双重催化能力,可以进行羟基化和有机底物去饱和。在这项研究中,我们采用密度泛函方法来探究该复合物观察到的非常高反应性的来源,并阐明该复合物如何有效地进行烷烃的羟基化和去饱和。通过对1可能参与反应机制的15 个可能的自旋态进行建模,我们的计算揭示了1的双重基态是由四重体 Fe IV中心和六重体 Fe III之间的反铁磁耦合引起的中心,它调节该物种的反应性。Fe IV O的高自旋基态异常稳定是由于与轨道的强烈重叠,通过自旋合作减少了反键相互作用。为1计算的电子结构特征与为所选方法提供信心的实验一致。此外,我们探索了 C-H 键活化以及烷烃的 -OH 反弹/去饱和的各种机制途径。为末端 Fe IV提取的第一个氢原子计算的极小的势垒高度O 单位被发现负责实验中观察到的百万倍激活。由 Fe III -OH 单元计算的 -OH 反弹的势垒高度也较小,表明有机底物容易羟基化1。两个铁中心之间的强自旋合作也降低了第二个氢原子提取的障碍,从而使去饱和路径具有竞争力。两个金属中心之间的自旋态和自旋耦合在决定物种1的反应性方面起着至关重要的作用. 通过探索各种机制途径,我们的研究揭示了桥接的 μ-氧代基团对于 C-H 活化和 -OH 反弹都是不良的亲电试剂。随着近年来越来越多的证据表明 sMMO 酶的开放核心几何形状,通过开放核心基序增强反应性的想法具有深远的影响。
更新日期:2020-06-18
O is due to the strong overlap of with the orbital, reducing the antibonding interactions via spin-cooperation. The electronic structure features computed for 1 are consistent with experiments offering confidence in the methodology chosen. Further, we have probed various mechanistic pathways for the C–H bond activation as well as –OH rebound/desaturation of alkanes. An extremely small barrier height computed for the first hydrogen atom abstraction by the terminal FeIVO unit was found to be responsible for the million-fold activation observed in the experiments. The barrier height computed for –OH rebound by the FeIII–OH unit is also smaller suggesting a facile hydroxylation of organic substrates by 1. A strong spin-cooperation between the two iron centres also reduces the barrier for second hydrogen atom abstraction, thus making the desaturation pathway competitive. Both the spin-state as well as spin-coupling between the two metal centres play a crucial role in dictating the reactivity for species 1. By exploring various mechanistic pathways, our study unveils the fact that the bridged μ-oxo group is a poor electrophile for both C–H activation as well for –OH rebound. As more and more evidence is gathered in recent years for the open core geometry of sMMO enzymes, the idea of enhancing the reactivity via an open-core motif has far-reaching consequences.
中文翻译:
破译开核二铁 [HO-FeIII-O-FeIVO]2+ 物种对 C-H 键活化观察到的百万倍反应性的起源:自旋态、自旋耦合和自旋合作的作用
高价金属-氧代物种已被表征为血红素和非血红素酶的关键中间体,被发现可进行有效的脂肪族羟基化、环氧化、卤化和脱氢反应。多年来已经合成了几种仿生模型复合物来模拟金属酶的结构和功能。金刚石核心 [Fe 2 (μ-O) 2 ] 是这方面著名的模型之一,因为它已被提议作为可溶性甲烷单加氧酶 (sMMO) 中的催化活性物质,它执行甲烷的具有挑战性的化学转化对甲醇放心。在此背景下,关于开核 [HO(L)Fe III –O–Fe IV (O)(L)] 2+ ( 1) 受到关注,因为与金刚石核心结构相比,它激活 C-H 键的速度快了一百万倍,并且具有双重催化能力,可以进行羟基化和有机底物去饱和。在这项研究中,我们采用密度泛函方法来探究该复合物观察到的非常高反应性的来源,并阐明该复合物如何有效地进行烷烃的羟基化和去饱和。通过对1可能参与反应机制的15 个可能的自旋态进行建模,我们的计算揭示了1的双重基态是由四重体 Fe IV中心和六重体 Fe III之间的反铁磁耦合引起的中心,它调节该物种的反应性。Fe IV O的高自旋基态异常稳定
是由于与轨道的强烈重叠,通过自旋合作减少了反键相互作用。为1计算的电子结构特征与为所选方法提供信心的实验一致。此外,我们探索了 C-H 键活化以及烷烃的 -OH 反弹/去饱和的各种机制途径。为末端 Fe IV提取的第一个氢原子计算的极小的势垒高度O 单位被发现负责实验中观察到的百万倍激活。由 Fe III -OH 单元计算的 -OH 反弹的势垒高度也较小,表明有机底物容易羟基化1。两个铁中心之间的强自旋合作也降低了第二个氢原子提取的障碍,从而使去饱和路径具有竞争力。两个金属中心之间的自旋态和自旋耦合在决定物种1的反应性方面起着至关重要的作用. 通过探索各种机制途径,我们的研究揭示了桥接的 μ-氧代基团对于 C-H 活化和 -OH 反弹都是不良的亲电试剂。随着近年来越来越多的证据表明 sMMO 酶的开放核心几何形状,通过开放核心基序增强反应性的想法具有深远的影响。

























 京公网安备 11010802027423号
京公网安备 11010802027423号