Computational and Structural Biotechnology Journal ( IF 6 ) Pub Date : 2020-06-18 , DOI: 10.1016/j.csbj.2020.06.020 Burak T Kaynak 1 , Ivet Bahar 1 , Pemra Doruker 1
|
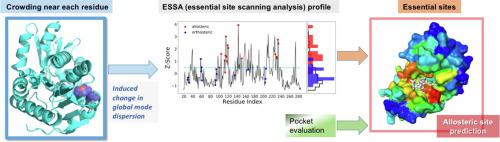
Despite the wealth of methods developed for exploring the molecular basis of allostery in biomolecular systems, there is still a need for structure-based predictive tools that can efficiently detect susceptible sites for triggering allosteric responses. Toward this goal, we introduce here an elastic network model (ENM)-based method, Essential Site Scanning Analysis (ESSA). Essential sites are here defined as residues that would significantly alter the protein’s global dynamics if bound to a ligand. To mimic the crowding induced upon substrate binding, the heavy atoms of each residue are incorporated as additional network nodes into the α-carbon-based ENM, and the resulting shifts in soft mode frequencies are used as a metric for evaluating the essentiality of each residue. Results on a dataset of monomeric proteins indicate the enrichment of allosteric and orthosteric binding sites, as well as global hinge regions among essential residues, highlighting the significant role of these sites in controlling the overall structural dynamics. Further integration of ESSA with information on predicted pockets and their local hydrophobicity density enables successful predictions of allosteric pockets for both ligand-bound and -unbound structures. ESSA can be efficiently applied to large multimeric systems. Three case studies, namely (i) G-protein binding to a GPCR, (ii) heterotrimeric assembly of the Ser/Thr protein phosphatase PP2A, and (iii) allo-targeting of AMPA receptor, demonstrate the utility of ESSA for identifying essential sites and narrowing down target allosteric sites identified by druggability simulations.
中文翻译:
基本位点扫描分析:一种检测调节蛋白质全局运动分散的位点的新方法。
尽管开发了丰富的方法来探索生物分子系统中变构的分子基础,但仍然需要基于结构的预测工具来有效检测易感位点以触发变构反应。为实现这一目标,我们在此介绍一种基于弹性网络模型 (ENM) 的方法,即基本站点扫描分析 (ESSA)。必要位点在这里被定义为如果与配体结合会显着改变蛋白质的全局动力学的残基。为了模拟底物结合时引起的拥挤,每个残基的重原子作为额外的网络节点合并到基于 α-碳的 ENM 中,并且由此产生的软模式频率的变化被用作评估每个残基的必要性的度量. 单体蛋白质数据集的结果表明变构和正构结合位点以及必需残基之间的全局铰链区的富集,突出了这些位点在控制整体结构动力学中的重要作用。将 ESSA 与有关预测口袋及其局部疏水密度的信息进一步整合,可以成功预测配体结合和未结合结构的变构口袋。ESSA 可以有效地应用于大型多聚体系统。三个案例研究,即 (i) G 蛋白与 GPCR 的结合,(ii) Ser/Thr 蛋白磷酸酶 PP2A 的异源三聚体组装,以及 (iii) AMPA 受体的同种异体靶向,证明了 ESSA 在识别必需位点方面的实用性并缩小通过成药性模拟确定的目标变构位点。


























 京公网安备 11010802027423号
京公网安备 11010802027423号