Computational and Theoretical Chemistry ( IF 2.8 ) Pub Date : 2020-06-17 , DOI: 10.1016/j.comptc.2020.112908 Muhammad Yasir Mehboob , Riaz Hussain , Muhammad Usman Khan , Muhammad Adnan , Ali Umar , Muhammad Usman Alvi , Mahmood Ahmed , Muhammad Khalid , Javed Iqbal , Muhammad Nadeem Akhtar , Fatiqa Zafar , Mahrzadi Noureen Shahi
|
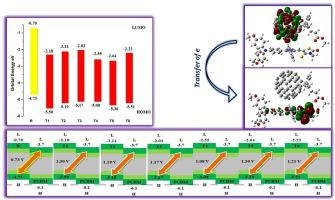
Considering the significant role of photovoltaic materials in diverse optoelectronic applications, N-phenylaniline-based six new donor molecules (T1-T6) are quantum chemically explored herein. Various parameters like frontier molecular orbital (FMO), density of states (DOS), transition density matrix (TDM) analysis, absorption maxima, reorganization energies of electron and hole, open circuit voltage (Voc), photophysical characteristics and charge transfer analysis have been estimated in order to understand the performance of newly designed molecules. End caped acceptors modification causes narrowing of HOMO-LUMO energy gap (2.99–3.33 eV) as compared to R (4.04 eV). All designed molecules exhibited absorption spectrum in the range of 334–370 nm in solvent phase and 324–403 nm in gas phase. T1-T6 show maximum charge transfer from HOMO to LUMO orbital which plays a key role in conductive materials. Dipole moment value of designed molecules in ground and excited states are found to be larger than reference molecule which suggested that our designed molecules have more solubility in organic solvent as compared to reference molecule. All designed molecules show better reorganizational energy of electron (0.0191–0.0273 Eh) and hole (0.0063–0.0187Eh). Among designed molecules T3 has lowest reorganizational energy of electron which means that it has highest charge mobility. The Voc with respect to HOMOdonor-LUMOPC61BM shows that designed molecules [T1-T6 (Voc = 1.08–1.50 V)] have better Voc as compared to R (Voc = 0.73 V). This theoretical framework proves that conceptualized molecules are superior and thus are recommended for the future construction of high performance organic solar cells.
中文翻译:
设计具有前途光电子性能的N-苯基苯胺-三唑构型供体材料,用于高效太阳能电池
考虑到光伏材料在各种光电应用中的重要作用,本文在量子化学上探索了基于N-苯基苯胺的六个新供体分子(T1-T6)。边界分子轨道(FMO),状态密度(DOS),跃迁密度矩阵(TDM)分析,吸收最大值,电子和空穴的重组能,开路电压(V oc),光物理特性和电荷转移分析等各种参数具有为了了解新设计分子的性能而进行了估算。与R相比,封端的受体修饰导致HOMO-LUMO能隙(2.99–3.33 eV)变窄(4.04 eV)。所有设计的分子在溶剂相中显示的吸收光谱范围为334–370 nm,在气相中显示为324–403 nm。T1-T6显示出从HOMO到LUMO轨道的最大电荷转移,这在导电材料中起关键作用。发现在基态和激发态下设计分子的偶极矩值大于参考分子,这表明我们设计的分子与参考分子相比在有机溶剂中的溶解度更高。所有设计的分子均显示出更好的电子(0.0191–0.0273 E h)和空穴(0.0063–0.0187E h)的重组能。在设计的分子中T3具有最低的电子重组能,这意味着它具有最高的电荷迁移率。相对于HOMO供体-LUMO PC61BM的V oc显示,与R(V oc = 0.73 V)相比,设计分子[ T1-T6(V oc = 1.08-1.50 V)]具有更好的V oc。该理论框架证明了概念化的分子是优越的,因此被推荐用于高性能有机太阳能电池的未来构造。

























 京公网安备 11010802027423号
京公网安备 11010802027423号