Bioorganic Chemistry ( IF 5.1 ) Pub Date : 2020-06-17 , DOI: 10.1016/j.bioorg.2020.104019 Abdallah Turky 1 , Ashraf H Bayoumi 1 , Adel Ghiaty 1 , Adel S El-Azab 2 , Alaa A-M Abdel-Aziz 2 , Hamada S Abulkhair 3
|
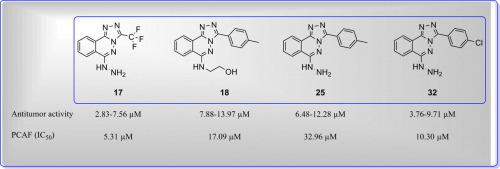
The antitumor activity of newly synthesised triazolophthalazines (L-45 analogues) 10–32 was evaluated in human hepatocellular carcinoma (HePG-2), breast cancer (MCF-7), prostate cancer (PC3), and colorectal carcinoma (HCT-116) cells. Compounds 17, 18, 25, and 32 showed potent antitumor activity (IC50, 2.83–13.97 μM), similar to doxorubicin (IC50, 4.17–8.87 μM) and afatinib (IC50, 5.4–11.4 μM). HePG2 was inhibited by compounds 10, 17, 18, 25, 26, and 32 (IC50, 3.06–10.5 μM), similar to doxorubicin (IC50, 4.50 μM) and afatinib (IC50, 5.4 μM). HCT-116 and MCF-7 were susceptible to compounds 10, 17, 18, 25, and 32 (IC50, 2.83–10.36 and 5.69–11.36 μM, respectively), similar to doxorubicin and afatinib (IC50 = 5.23 and 4.17, and 11.4 and 7.1 μM, respectively). Compounds 17, 25, and 32 exerted potent activities against PC3 (IC50, 7.56–12.28 μM) compared with doxorubicin (IC50, 8.87 µM) and afatinib (IC50 7.7 μM). Compounds 17 and 32 were the strongest PCAF inhibitors (IC50, 5.31 and 10.30 μM, respectively) and compounds 18 and 25 exhibited modest IC50 values (17.09 and 32.96 μM, respectively) compared with bromosporine (IC50, 5.00 μM). Compound 17 was cytotoxic to HePG2 cells (IC50, 3.06 μM), inducing apoptosis in the pre-G phase and arresting the cell cycle in the G2/M phase. Molecular docking for the most active PCAF inhibitors (17 and 32) was performed.
中文翻译:
基于1,2,4-三唑并酞嗪支架的新型化合物的设计,合成和抗肿瘤活性:诱导细胞凋亡和抑制PCAF。
在人肝细胞癌(HePG-2),乳腺癌(MCF-7),前列腺癌(PC3)和结直肠癌(HCT-116)中评估了新合成的三唑酞嗪(L-45类似物)10 – 32的抗肿瘤活性。细胞。化合物17,18,25,和32显示出强效的抗肿瘤活性(IC 50,μM2.83-13.97),类似于多柔比星(IC 50,4.17-8.87μM)和阿法(IC 50,μM5.4-11.4)。的HePG2通过化合物抑制10,17,18,25,26,和32(IC 50,3.06-10.5μM),类似于多柔比星(IC 50,μM4.50)和阿法(IC 50,μM5.4)。HCT-116和MCF-7均对化合物10,17,18,25,和32(IC 50,2.83-10.36和5.69-11.36μM,分别地),类似于阿霉素和阿法(IC 50 = 5.23和4.17,分别为11.4和7.1μM)。化合物17,25和32个施加的对抗PC3(IC有力活动50与阿霉素相比,7.56-12.28μM)(IC 50,8.87μM)和阿法(IC 507.7μM)。化合物17和32分别为最强PCAF抑制剂(IC 50,5.31和10.30微米,分别地)和化合物18个25表现出适度的IC 50值(17.09和32.96μM,分别地)与bromosporine比较(IC 50,μM5.00)。化合物17对HePG2细胞具有细胞毒性(IC 50为3.06μM),在pre-G期诱导细胞凋亡,并在G2 / M期阻止细胞周期。进行了对最具活性的PCAF抑制剂(17和32)的分子对接。

























 京公网安备 11010802027423号
京公网安备 11010802027423号