Computational and Structural Biotechnology Journal ( IF 6 ) Pub Date : 2020-06-12 , DOI: 10.1016/j.csbj.2020.06.012 Seungbyn Baek 1 , Insuk Lee 1, 2
|
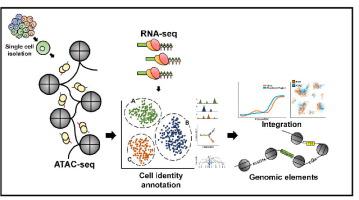
Most genetic variations associated with human complex traits are located in non-coding genomic regions. Therefore, understanding the genotype-to-phenotype axis requires a comprehensive catalog of functional non-coding genomic elements, most of which are involved in epigenetic regulation of gene expression. Genome-wide maps of open chromatin regions can facilitate functional analysis of cis- and trans-regulatory elements via their connections with trait-associated sequence variants. Currently, Assay for Transposase Accessible Chromatin with high-throughput sequencing (ATAC-seq) is considered the most accessible and cost-effective strategy for genome-wide profiling of chromatin accessibility. Single-cell ATAC-seq (scATAC-seq) technology has also been developed to study cell type-specific chromatin accessibility in tissue samples containing a heterogeneous cellular population. However, due to the intrinsic nature of scATAC-seq data, which are highly noise and sparse, accurate extraction of biological signals and devising effective biological hypothesis is difficult. To overcome such limitations in scATAC-seq data analysis, new methods and software tools have been developed over the past few years. Nevertheless, there is no consensus for the best practice of scATAC-seq data analysis yet. In this review, we discuss scATAC-seq technology and data analysis methods, ranging from preprocessing to downstream analysis, along with an up-to-date list of published studies that involved the application of this method. We expect this review will provide a guideline for successful data generation and analysis methods using appropriate software tools and databases for the study of chromatin accessibility at single-cell resolution.
中文翻译:
单细胞 ATAC 测序分析:从数据预处理到假设生成。
大多数与人类复杂性状相关的遗传变异都位于非编码基因组区域。因此,了解基因型到表型轴需要功能性非编码基因组元件的全面目录,其中大多数涉及基因表达的表观遗传调控。开放染色质区域的全基因组图谱可以通过顺式和反式调控元件与性状相关序列变体的连接来促进顺式和反式调控元件的功能分析。目前,通过高通量测序 (ATAC-seq) 检测转座酶可及染色质被认为是全基因组染色质可及性分析最容易实现且最具成本效益的策略。单细胞 ATAC-seq (scATAC-seq) 技术也被开发用于研究含有异质细胞群的组织样本中细胞类型特异性染色质的可及性。然而,由于 scATAC-seq 数据的固有性质,即高噪声和稀疏,准确提取生物信号并设计有效的生物学假设很困难。为了克服 scATAC-seq 数据分析中的此类限制,过去几年开发了新的方法和软件工具。然而,对于 scATAC-seq 数据分析的最佳实践尚未达成共识。在这篇综述中,我们讨论了 scATAC-seq 技术和数据分析方法,从预处理到下游分析,以及涉及该方法应用的最新已发表研究列表。我们期望这篇综述将为使用适当的软件工具和数据库成功地生成数据和分析方法提供指导,以研究单细胞分辨率的染色质可及性。


























 京公网安备 11010802027423号
京公网安备 11010802027423号