当前位置:
X-MOL 学术
›
Chem. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Hardware efficient quantum algorithms for vibrational structure calculations.
Chemical Science ( IF 8.4 ) Pub Date : 2020-06-11 , DOI: 10.1039/d0sc01908a Pauline J Ollitrault 1, 2 , Alberto Baiardi 2 , Markus Reiher 2 , Ivano Tavernelli 1
Chemical Science ( IF 8.4 ) Pub Date : 2020-06-11 , DOI: 10.1039/d0sc01908a Pauline J Ollitrault 1, 2 , Alberto Baiardi 2 , Markus Reiher 2 , Ivano Tavernelli 1
Affiliation
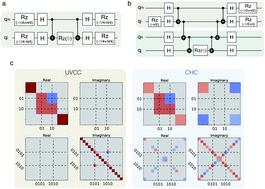
|
We introduce a framework for the calculation of ground and excited state energies of bosonic systems suitable for near-term quantum devices and apply it to molecular vibrational anharmonic Hamiltonians. Our method supports generic reference modal bases and Hamiltonian representations, including the ones that are routinely used in classical vibrational structure calculations. We test different parametrizations of the vibrational wavefunction, which can be encoded in quantum hardware, based either on heuristic circuits or on the bosonic Unitary Coupled Cluster Ansatz. In particular, we define a novel compact heuristic circuit and demonstrate that it provides a good compromise in terms of circuit depth, optimization costs, and accuracy. We evaluate the requirements, number of qubits and circuit depth, for the calculation of vibrational energies on quantum hardware and compare them with state-of-the-art classical vibrational structure algorithms for molecules with up to seven atoms.
中文翻译:

用于振动结构计算的硬件高效量子算法。
我们引入了一个计算适用于近期量子器件的玻色子系统的基态和激发态能量的框架,并将其应用于分子振动非谐哈密顿量。我们的方法支持通用参考模态基础和哈密顿表示,包括经典振动结构计算中常规使用的那些。我们测试了振动波函数的不同参数化,这些参数化可以基于启发式电路或玻色酉耦合簇Ansatz在量子硬件中进行编码。特别是,我们定义了一种新颖的紧凑启发式电路,并证明它在电路深度、优化成本和准确性方面提供了良好的折衷。我们评估了量子硬件上振动能量计算的要求、量子位数量和电路深度,并将其与最多七个原子的分子的最先进的经典振动结构算法进行比较。
更新日期:2020-07-08
中文翻译:
用于振动结构计算的硬件高效量子算法。
我们引入了一个计算适用于近期量子器件的玻色子系统的基态和激发态能量的框架,并将其应用于分子振动非谐哈密顿量。我们的方法支持通用参考模态基础和哈密顿表示,包括经典振动结构计算中常规使用的那些。我们测试了振动波函数的不同参数化,这些参数化可以基于启发式电路或玻色酉耦合簇Ansatz在量子硬件中进行编码。特别是,我们定义了一种新颖的紧凑启发式电路,并证明它在电路深度、优化成本和准确性方面提供了良好的折衷。我们评估了量子硬件上振动能量计算的要求、量子位数量和电路深度,并将其与最多七个原子的分子的最先进的经典振动结构算法进行比较。


























 京公网安备 11010802027423号
京公网安备 11010802027423号