School of Chemical Engineering, Nanjing University of Science and Technology, Nanjing, 210094, China.
A series of furazan substituted s-triazine derivatives were designed and investigated theoretically as potential nitrogen-rich high-energy-density materials in this work. Density functional theory (DFT) methods were used to predict the heats of formation (HOFs) and compounds structure was optimized at B3PW91/6-31G++ (d,p) level. The explosive detonation parameters were calculated based on Kamlet−Jacobs equations and Born−Haber cycle. The presence of the −NO2 and − NH2 groups in the same structure were found to be helpful in improving structural stability through intramolecular weak interactions. Most of the designed compounds were characterized by high HOFs (solid-phase heat of information 71.01–518.20 kJ/mol) and crystal density values (1.74–1.90 g/cm3). In the analysis of frontier molecular orbital that some designed compounds chemical activity similar with TATB, but show better detonation performance. The predicted results reveal that some designed nitrogen-rich compounds outperform traditional energetic materials and may be considered as potential candidates for high-energy materials.
BRIEFS A series of furazan substituted s-triazine derivatives were designed and investigated theoretically as potential nitrogen-rich high-energy-density materials and most of the compounds exhibit high solid phase heat of information and fascinating detonation properties
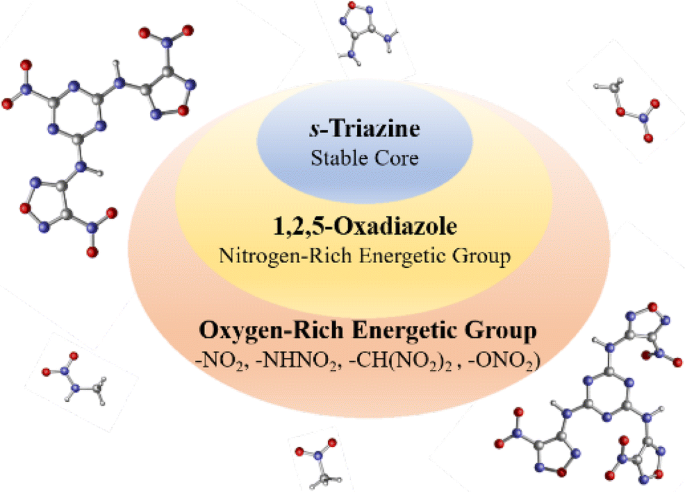 BRIEFS A series of furazan substituted s-triazine derivatives were designed and investigated theoretically as potential nitrogen-rich high-energy-density materials and most of the compounds exhibit high solid phase heat of information and fascinating detonation properties
BRIEFS A series of furazan substituted s-triazine derivatives were designed and investigated theoretically as potential nitrogen-rich high-energy-density materials and most of the compounds exhibit high solid phase heat of information and fascinating detonation properties
 BRIEFS A series of furazan substituted s-triazine derivatives were designed and investigated theoretically as potential nitrogen-rich high-energy-density materials and most of the compounds exhibit high solid phase heat of information and fascinating detonation properties
BRIEFS A series of furazan substituted s-triazine derivatives were designed and investigated theoretically as potential nitrogen-rich high-energy-density materials and most of the compounds exhibit high solid phase heat of information and fascinating detonation properties
























 京公网安备 11010802027423号
京公网安备 11010802027423号