当前位置:
X-MOL 学术
›
Acta Cryst. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Charge density of 4-methyl-3-[(tetrahydro-2H-pyran-2-yl)oxy]thiazole-2(3H)-thione. A comprehensive multipole refinement, maximum entropy method and density functional theory study.
Acta Crystallographica Section B ( IF 2.684 ) Pub Date : 2020-06-08 , DOI: 10.1107/s2052520620005533 Barbora Vénosová 1 , Julia Koziskova 1 , Jozef Kožíšek 1 , Peter Herich 1 , Karol Lušpai 1 , Vaclav Petricek 2 , Jens Hartung 3 , Mike Müller 3 , Christian B Hübschle 4 , Sander van Smaalen 4 , Lukas Bucinsky 1
Acta Crystallographica Section B ( IF 2.684 ) Pub Date : 2020-06-08 , DOI: 10.1107/s2052520620005533 Barbora Vénosová 1 , Julia Koziskova 1 , Jozef Kožíšek 1 , Peter Herich 1 , Karol Lušpai 1 , Vaclav Petricek 2 , Jens Hartung 3 , Mike Müller 3 , Christian B Hübschle 4 , Sander van Smaalen 4 , Lukas Bucinsky 1
Affiliation
|
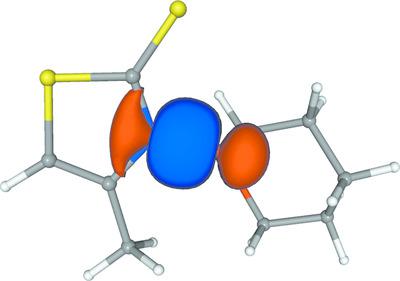
The structure of 4‐methyl‐3‐[(tetrahydro‐2H‐pyran‐2‐yl)oxy]thiazole‐2(3H)‐thione (MTTOTHP) was investigated using X‐ray diffraction and computational chemistry methods for determining properties of the nitrogen—oxygen bond, which is the least stable entity upon photochemical excitation. Experimentally measured structure factors have been used to determine and characterize charge density via the multipole model (MM) and the maximum entropy method (MEM). Theoretical investigation of the electron density and the electronic structure has been performed in the finite basis set density functional theory (DFT) framework. Quantum Theory of Atoms In Molecules (QTAIM), deformation densities and Laplacians maps have been used to compare theoretical and experimental results. MM experimental results and predictions from theory differ with respect to the sign and/or magnitude of the Laplacian at the N—O bond critical point (BCP), depending on the treatment of n values of the MM radial functions. Such Laplacian differences in the N—O bond case are discussed with respect to a lack of flexibility in the MM radial functions also reported by Rykounov et al. [Acta Cryst. (2011), B67, 425–436]. BCP Hessian eigenvalues show qualitatively matching results between MM and DFT. In addition, the theoretical analysis used domain‐averaged fermi holes (DAFH), natural bond orbital (NBO) analysis and localized (LOC) orbitals to characterize the N—O bond as a single σ bond with marginal π character. Hirshfeld atom refinement (HAR) has been employed to compare to the MM refinement results and/or neutron dataset C—H bond lengths and to crystal or single molecule geometry optimizations, including considerations of anisotropy of H atoms. Our findings help to understand properties of molecules like MTTOTHP as progenitors of free oxygen radicals.
中文翻译:

4-甲基-3-[(四氢-2H-吡喃-2-基)氧基]噻唑-2(3H)-硫酮的电荷密度。全面的多极细化,最大熵方法和密度泛函理论研究。
4-甲基-3-[(四氢-2 H-吡喃-2-基)氧基]噻唑-2(3 H)-硫酮的结构(MTTOTHP)是使用X射线衍射和计算机化学方法研究的,用于确定氮-氧键的性质,该氮-氧键是光化学激发后最不稳定的实体。实验测量的结构因子已通过多极模型(MM)和最大熵方法(MEM)用于确定和表征电荷密度。电子密度和电子结构的理论研究已在有限基集密度泛函理论(DFT)框架中进行。分子中的原子量子理论(QTAIM),变形密度和拉普拉斯图已经用于比较理论和实验结果。MM实验结果和理论预测在N键键临界点(BCP)上的拉普拉斯算符和/或大小不同,MM径向函数的n个值。Rykounov等人也报道了在MM径向功能缺乏灵活性方面讨论了这种N-O键情况下的拉普拉斯差异。[ Acta Cryst。(2011),B 67,425–436]。BCP Hessian特征值显示MM和DFT之间的定性匹配结果。此外,理论分析还使用了域平均费米空穴(DAFH),自然键轨道(NBO)分析和局部(LOC)轨道,将N-O键表征为具有边际π特征的单个σ键。Hirshfeld原子精炼(HAR)已用于与MM精炼结果和/或中子数据集CH键长度进行比较,并与晶体或单分子几何优化(包括考虑H原子的各向异性)进行比较。我们的发现有助于理解像MTTOTHP这样的分子作为游离氧自由基的祖先的性质。
更新日期:2020-06-08
中文翻译:
4-甲基-3-[(四氢-2H-吡喃-2-基)氧基]噻唑-2(3H)-硫酮的电荷密度。全面的多极细化,最大熵方法和密度泛函理论研究。
4-甲基-3-[(四氢-2 H-吡喃-2-基)氧基]噻唑-2(3 H)-硫酮的结构(MTTOTHP)是使用X射线衍射和计算机化学方法研究的,用于确定氮-氧键的性质,该氮-氧键是光化学激发后最不稳定的实体。实验测量的结构因子已通过多极模型(MM)和最大熵方法(MEM)用于确定和表征电荷密度。电子密度和电子结构的理论研究已在有限基集密度泛函理论(DFT)框架中进行。分子中的原子量子理论(QTAIM),变形密度和拉普拉斯图已经用于比较理论和实验结果。MM实验结果和理论预测在N键键临界点(BCP)上的拉普拉斯算符和/或大小不同,MM径向函数的n个值。Rykounov等人也报道了在MM径向功能缺乏灵活性方面讨论了这种N-O键情况下的拉普拉斯差异。[ Acta Cryst。(2011),B 67,425–436]。BCP Hessian特征值显示MM和DFT之间的定性匹配结果。此外,理论分析还使用了域平均费米空穴(DAFH),自然键轨道(NBO)分析和局部(LOC)轨道,将N-O键表征为具有边际π特征的单个σ键。Hirshfeld原子精炼(HAR)已用于与MM精炼结果和/或中子数据集CH键长度进行比较,并与晶体或单分子几何优化(包括考虑H原子的各向异性)进行比较。我们的发现有助于理解像MTTOTHP这样的分子作为游离氧自由基的祖先的性质。

























 京公网安备 11010802027423号
京公网安备 11010802027423号