Current Computer-Aided Drug Design ( IF 1.7 ) Pub Date : 2020-05-31 , DOI: 10.2174/1573409915666191010104527 Thomas Scior 1 , Hassan H Abdallah 2, 3 , Kenia Salvador-Atonal 1 , Stefan Laufer 4
|
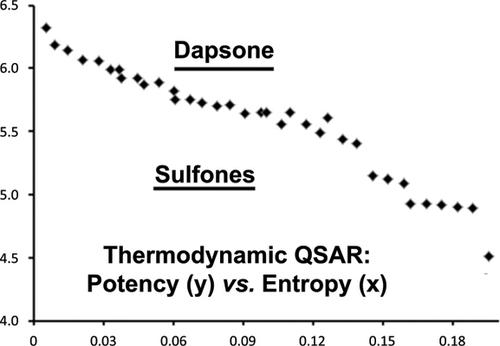
Background: The relatedness between the linear equations of thermodynamics and QSAR was studied thanks to the recently elucidated crystal structure complexes between sulfonamide pterin conjugates and dihydropteroate synthase (DHPS) together with a published set of thirty- six synthetic dapsone derivatives with their reported entropy-driven activity data. Only a few congeners were slightly better than dapsone.
Objective: Our study aimed at demonstrating the applicability of thermodynamic QSAR and to shed light on the mechanistic aspects of sulfone binding to DHPS.
Methods: To this end ligand docking to DHPS, quantum mechanical properties, 2D- and 3D-QSAR as well as Principle Component Analysis (PCA) were carried out.
Results: The short aryl substituents of the docked pterin-sulfa conjugates were outward oriented into the solvent space without interacting with target residues which explains why binding enthalpy (ΔH) did not correlate with potency. PCA revealed how chemically informative descriptors are evenly loaded on the first three PCs (interpreted as ΔG, ΔH and ΔS), while chemically cryptic ones reflected higher dimensional (complex) loadings.
Conclusion: It is safe to utter that synthesis efforts to introduce short side chains for aryl derivatization of the dapsone scaffold have failed in the past. On theoretical grounds we provide computed evidence why dapsone is not a pharmacodynamic lead for drug profiling because enthalpic terms do not change significantly at the moment of ligand binding to target.
中文翻译:
氨苯砜不是其芳基衍生物的药效先导化合物。
背景:由于最近阐明了磺酰胺蝶呤共轭物和二氢蝶呤合酶(DHPS)之间的晶体结构复合物,以及已发表的一组三十六种合成的氨苯砜衍生物,并据报道由熵驱动,因此研究了热力学与QSAR线性方程之间的相关性。活动数据。只有少数同类产品略优于氨苯砜。
目的:我们的研究旨在证明热力学QSAR的适用性,并阐明砜与DHPS结合的机理。
方法:为此,配体与DHPS对接,进行了量子力学性能,2D和3D-QSAR以及主成分分析(PCA)。
结果:对接的蝶呤-磺基共轭物的短芳基取代基向外定向到溶剂空间中,而没有与目标残基相互作用,这解释了为什么结合焓(ΔH)与效能不相关。PCA揭示了如何在前三台PC上均匀地加载化学信息描述符(解释为ΔG,ΔH和ΔS),而化学隐秘的描述符则反映了更高维度(复杂)的载荷。
结论:可以肯定地说,过去引入氨苯砜骨架芳基衍生化短侧链的合成努力失败了。从理论上讲,我们提供了计算证据,证明氨苯砜为什么不是药物谱分析的药效学先导,因为在配体与靶标结合时,焓项没有显着变化。


























 京公网安备 11010802027423号
京公网安备 11010802027423号