Biochimica et Biophysica Acta (BBA) - General Subjects ( IF 3 ) Pub Date : 2020-06-05 , DOI: 10.1016/j.bbagen.2020.129653 Mehmet Ali Öztürk 1 , Rebecca C Wade 2
|
Background
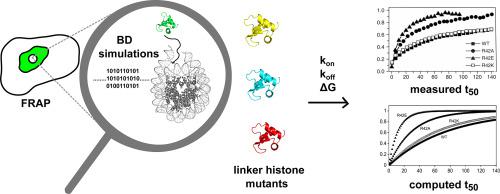
Fluorescence recovery after photobleaching (FRAP) studies can provide kinetic information about proteins in cells. Single point mutations can significantly affect the binding kinetics of proteins and result in variations in the recovery half time (t50) measured in FRAP experiments. FRAP measurements of linker histone (LH) proteins in the cell nucleus have previously been reported by Brown et al. (2006) and Lele et al. (2006).
Methods
We performed Brownian dynamics (BD) simulations of the diffusional association of the wild-type and 38 single or double point mutants of the globular domain of mouse linker histone H1.0 (gH1.0) to a nucleosome. From these simulations, we calculated the bimolecular association rate constant (kon), the Gibbs binding free energy (ΔG) and the dissociation rate constant (koff) related to formation of a diffusional encounter complex between the nucleosome and the gH1.0.
Results
We used these parameters, after application of a correction factor to account for the effects of the crowded environment of the nucleus, to compute FRAP recovery times and curves that are in good agreement with previously published, experimentally measured FRAP recovery time courses.
Conclusions
Our computational analysis suggests that BD simulations can be used to predict the relative effects of single point mutations on FRAP recovery times related to protein binding.
General Significance
BD simulations assist in providing a detailed molecular level interpretation of FRAP data.
中文翻译:
基于布朗动力学模拟计算接头组蛋白-染色质结合的FRAP恢复时间。
背景
光漂白后的荧光恢复(FRAP)研究可以提供有关细胞中蛋白质的动力学信息。单点突变会显着影响蛋白质的结合动力学,并导致在FRAP实验中测得的恢复半衰期(t 50)发生变化。Brown等人先前已经报道了细胞核中连接组蛋白(LH)蛋白的FRAP测量。(2006)和Lele等。(2006)。
方法
我们对小鼠接头组蛋白H1.0(gH1.0)的球状结构域的野生型和38个单点或双点突变体的扩散关联进行了核动力学(BD)模拟。从这些模拟中,我们计算了与核小体和gH1.0之间形成的扩散相遇复合物有关的双分子缔合速率常数(k on),吉布斯结合自由能(ΔG)和解离速率常数(k off)。
结果
在应用校正因子解决核拥挤环境的影响后,我们使用了这些参数来计算FRAP恢复时间和曲线,这些曲线与先前发布的,实验测量的FRAP恢复时间过程非常吻合。
结论
我们的计算分析表明,BD模拟可用于预测单点突变对与蛋白质结合相关的FRAP恢复时间的相对影响。
一般意义
BD模拟有助于提供FRAP数据的详细分子水平解释。

























 京公网安备 11010802027423号
京公网安备 11010802027423号