当前位置:
X-MOL 学术
›
Mol. Pharmaceutics
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Machine Estimation of Drug Melting Properties and Influence on Solubility Prediction.
Molecular Pharmaceutics ( IF 4.9 ) Pub Date : 2020-06-04 , DOI: 10.1021/acs.molpharmaceut.0c00355 Nicole Wyttenbach 1 , Andreas Niederquell 2 , Martin Kuentz 2
Molecular Pharmaceutics ( IF 4.9 ) Pub Date : 2020-06-04 , DOI: 10.1021/acs.molpharmaceut.0c00355 Nicole Wyttenbach 1 , Andreas Niederquell 2 , Martin Kuentz 2
Affiliation
|
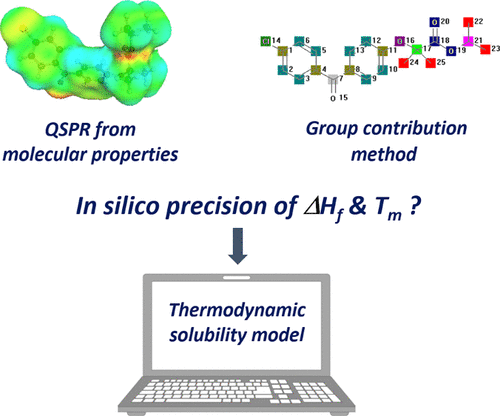
There has been much recent interest in machine learning (ML) and molecular quantitative structure property relationships (QSPR). The present research evaluated modern ML-based methods implemented in commercial software (COSMOquick and Molecular Modeling Pro), compared to a classical group contribution approach (Joback and Reid method), to estimate melting points and enthalpy of fusion values. A broad data set of market compounds was gathered from the literature, together with new data measured by differential scanning calorimetry for drug candidates. The highest prediction accuracy was achieved by QSPR using stochastic gradient boosting. The model deviations were discussed, particularly the implications on thermodynamic solubility modeling, as this typically requires estimation of both melting point and enthalpy of fusion. The results suggested that despite considerable advancement in prediction accuracy, there are still limitations especially with complex drug candidates. It is recommended that in such cases, melting properties obtained in silico should be used carefully as input data for thermodynamic solubility modeling. Future research will show how the prediction limits of thermophysical drug properties can be further advanced by even larger data sets and other ML algorithms or also by using molecular simulations.
中文翻译:

药物熔解特性的机器估计及其对溶解度预测的影响。
最近人们对机器学习(ML)和分子定量结构特性关系(QSPR)产生了浓厚的兴趣。本研究评估了在商业软件(COSMOquick和Molecular Modeling Pro)中实现的基于ML的现代方法,与传统的基团贡献方法(Joback和Reid方法)相比,可估计熔点和熔融焓。从文献中收集了广泛的市场化合物数据,以及通过差示扫描量热法对候选药物进行了测量的新数据。使用随机梯度提升的QSPR可以实现最高的预测精度。讨论了模型偏差,尤其是对热力学溶解度建模的影响,因为这通常需要估计熔点和熔融焓。结果表明,尽管预测准确性有了很大的提高,但仍然存在局限性,尤其是对于复杂的候选药物。建议在这种情况下,获得熔融性能应谨慎使用in silico作为热力学溶解度建模的输入数据。未来的研究将显示如何通过更大的数据集和其他ML算法或通过使用分子模拟来进一步提高热物理药物特性的预测极限。
更新日期:2020-07-06
中文翻译:
药物熔解特性的机器估计及其对溶解度预测的影响。
最近人们对机器学习(ML)和分子定量结构特性关系(QSPR)产生了浓厚的兴趣。本研究评估了在商业软件(COSMOquick和Molecular Modeling Pro)中实现的基于ML的现代方法,与传统的基团贡献方法(Joback和Reid方法)相比,可估计熔点和熔融焓。从文献中收集了广泛的市场化合物数据,以及通过差示扫描量热法对候选药物进行了测量的新数据。使用随机梯度提升的QSPR可以实现最高的预测精度。讨论了模型偏差,尤其是对热力学溶解度建模的影响,因为这通常需要估计熔点和熔融焓。结果表明,尽管预测准确性有了很大的提高,但仍然存在局限性,尤其是对于复杂的候选药物。建议在这种情况下,获得熔融性能应谨慎使用in silico作为热力学溶解度建模的输入数据。未来的研究将显示如何通过更大的数据集和其他ML算法或通过使用分子模拟来进一步提高热物理药物特性的预测极限。


























 京公网安备 11010802027423号
京公网安备 11010802027423号