当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Charge and Exciton Transfer Simulations Using Machine-Learned Hamiltonians.
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2020-06-03 , DOI: 10.1021/acs.jctc.0c00246 Mila Krämer 1, 2 , Philipp M Dohmen 1, 2 , Weiwei Xie 1 , Daniel Holub 1 , Anders S Christensen 3 , Marcus Elstner 1, 2
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2020-06-03 , DOI: 10.1021/acs.jctc.0c00246 Mila Krämer 1, 2 , Philipp M Dohmen 1, 2 , Weiwei Xie 1 , Daniel Holub 1 , Anders S Christensen 3 , Marcus Elstner 1, 2
Affiliation
|
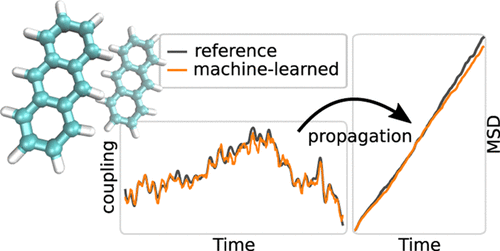
Quantum-mechanical simulations of charge and exciton transfer in molecular organic materials are a key method to increase our understanding of organic semiconductors. Our goal is to build an efficient multiscale model to predict charge-transfer mobilities and exciton diffusion constants from nonadiabatic molecular dynamics simulations and Marcus-based Monte Carlo approaches. In this work, we apply machine learning models to simulate charge and exciton propagation in organic semiconductors. We show that kernel ridge regression models can be trained to predict electronic and excitonic couplings from semiempirical density functional tight binding (DFTB) reference data with very good accuracy. In simulations, the models could reproduce hole mobilities along the anthracene crystal axes to within 8.5% of the DFTB reference and 34% of the experimental results with only 1000 training data points. Using these models decreased the cost of exciton transfer simulations by one order of magnitude.
中文翻译:

使用机器学习的哈密顿量进行电荷和激子传递模拟。
分子有机材料中电荷和激子转移的量子力学模拟是增加我们对有机半导体理解的一种关键方法。我们的目标是从非绝热分子动力学模拟和基于Marcus的蒙特卡洛方法建立一个高效的多尺度模型,以预测电荷转移迁移率和激子扩散常数。在这项工作中,我们应用机器学习模型来模拟有机半导体中的电荷和激子传播。我们显示,可以训练内核岭回归模型来预测来自半经验密度泛函紧密结合(DFTB)参考数据的电子和激子耦合,并且具有非常好的准确性。在模拟中,这些模型可以将沿蒽晶体轴的空穴迁移率复制到8以内。仅有1000个训练数据点的DFTB参考的5%和实验结果的34%。使用这些模型可以将激子转移模拟的成本降低一个数量级。
更新日期:2020-07-14
中文翻译:
使用机器学习的哈密顿量进行电荷和激子传递模拟。
分子有机材料中电荷和激子转移的量子力学模拟是增加我们对有机半导体理解的一种关键方法。我们的目标是从非绝热分子动力学模拟和基于Marcus的蒙特卡洛方法建立一个高效的多尺度模型,以预测电荷转移迁移率和激子扩散常数。在这项工作中,我们应用机器学习模型来模拟有机半导体中的电荷和激子传播。我们显示,可以训练内核岭回归模型来预测来自半经验密度泛函紧密结合(DFTB)参考数据的电子和激子耦合,并且具有非常好的准确性。在模拟中,这些模型可以将沿蒽晶体轴的空穴迁移率复制到8以内。仅有1000个训练数据点的DFTB参考的5%和实验结果的34%。使用这些模型可以将激子转移模拟的成本降低一个数量级。


























 京公网安备 11010802027423号
京公网安备 11010802027423号