Computational and Structural Biotechnology Journal ( IF 6 ) Pub Date : 2020-06-03 , DOI: 10.1016/j.csbj.2020.05.021 Jocelyn Gal 1 , Caroline Bailleux 2 , David Chardin 3, 4 , Thierry Pourcher 4 , Julia Gilhodes 5 , Lun Jing 4 , Jean-Marie Guigonis 4 , Jean-Marc Ferrero 2 , Gerard Milano 6 , Baharia Mograbi 7 , Patrick Brest 7 , Yann Chateau 1 , Olivier Humbert 3, 4 , Emmanuel Chamorey 1
|
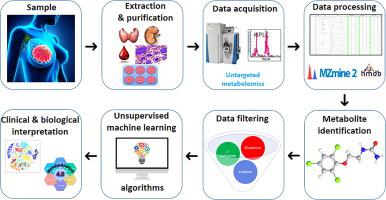
Genomics and transcriptomics have led to the widely-used molecular classification of breast cancer (BC). However, heterogeneous biological behaviors persist within breast cancer subtypes. Metabolomics is a rapidly-expanding field of study dedicated to cellular metabolisms affected by the environment. The aim of this study was to compare metabolomic signatures of BC obtained by 5 different unsupervised machine learning (ML) methods. Fifty-two consecutive patients with BC with an indication for adjuvant chemotherapy between 2013 and 2016 were retrospectively included. We performed metabolomic profiling of tumor resection samples using liquid chromatography-mass spectrometry. Here, four hundred and forty-nine identified metabolites were selected for further analysis. Clusters obtained using 5 unsupervised ML methods (PCA k-means, sparse k-means, spectral clustering, SIMLR and k-sparse) were compared in terms of clinical and biological characteristics. With an optimal partitioning parameter k = 3, the five methods identified three prognosis groups of patients (favorable, intermediate, unfavorable) with different clinical and biological profiles. SIMLR and K-sparse methods were the most effective techniques in terms of clustering. In-silico survival analysis revealed a significant difference for 5-year predicted OS between the 3 clusters. Further pathway analysis using the 449 selected metabolites showed significant differences in amino acid and glucose metabolism between BC histologic subtypes. Our results provide proof-of-concept for the use of unsupervised ML metabolomics enabling stratification and personalized management of BC patients. The design of novel computational methods incorporating ML and bioinformatics techniques should make available tools particularly suited to improving the outcome of cancer treatment and reducing cancer-related mortalities.
中文翻译:
用于识别局部乳腺癌患者代谢组学特征的无监督机器学习方法的比较。
基因组学和转录组学导致了乳腺癌 (BC) 的广泛使用的分子分类。然而,乳腺癌亚型中仍然存在异质的生物学行为。代谢组学是一个快速发展的研究领域,致力于研究受环境影响的细胞代谢。本研究的目的是比较通过 5 种不同的无监督机器学习 (ML) 方法获得的 BC 代谢组学特征。回顾性纳入 2013 年至 2016 年期间有辅助化疗指征的 52 名连续 BC 患者。我们使用液相色谱-质谱法对肿瘤切除样本进行代谢组学分析。在这里,选择了四百四十九种已鉴定的代谢物进行进一步分析。使用 5 种无监督 ML 方法(PCA k-means、稀疏 k-means、谱聚类、SIMLR 和 k-sparse)获得的聚类在临床和生物学特征方面进行了比较。采用最佳划分参数 k = 3,五种方法确定了具有不同临床和生物学特征的三组患者预后(良好、中间、不良)。SIMLR 和 K-稀疏方法是聚类方面最有效的技术。计算机生存分析显示,3 个集群之间的 5 年预测 OS 存在显着差异。使用 449 种选定代谢物进行的进一步通路分析显示 BC 组织学亚型之间的氨基酸和葡萄糖代谢存在显着差异。我们的结果为使用无监督 ML 代谢组学实现 BC 患者的分层和个性化管理提供了概念验证。结合机器学习和生物信息学技术的新型计算方法的设计应该提供特别适合改善癌症治疗结果和降低癌症相关死亡率的工具。


























 京公网安备 11010802027423号
京公网安备 11010802027423号