当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Extrapolating Unconverged GW Energies up to the Complete Basis Set Limit with Linear Regression.
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2020-06-03 , DOI: 10.1021/acs.jctc.0c00433 Fabien Bruneval 1 , Ivan Maliyov 1 , Clovis Lapointe 1 , Mihai-Cosmin Marinica 1
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2020-06-03 , DOI: 10.1021/acs.jctc.0c00433 Fabien Bruneval 1 , Ivan Maliyov 1 , Clovis Lapointe 1 , Mihai-Cosmin Marinica 1
Affiliation
|
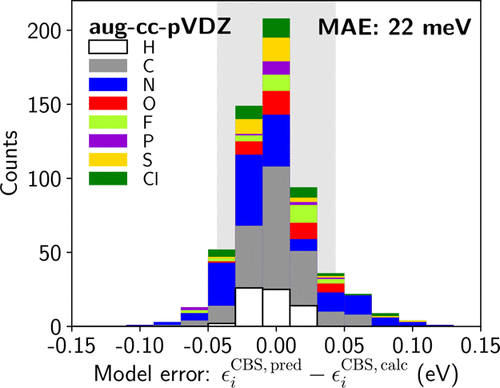
The GW approximation to the electronic self-energy is now a well-recognized approach to obtain the electron quasiparticle energies of molecules and, in particular, their ionization potential and electron affinity. Though much faster than the corresponding wavefunction methods, the GW energies are still affected by slow convergence with respect to the basis completeness. This limitation hinders a wider application of the GW approach. Here, we show that we can reach the complete basis set limit for the cumbersome GW calculations solely based on fast preliminary calculations with an unconverged basis set. We introduce a linear model that correlates the molecular orbital characteristics and the basis convergence error for a large database of approximately 600 states in 104 organic molecules that contain H, C, O, N, F, P, S, and Cl. The model employs molecular-orbital-based non-linear descriptors that encode efficiently the chemical space offering outstanding transferability. Using a low number of descriptors (17) the performance of this extrapolation procedure is superior to that of the earlier more physically motivated approaches. The predictive power of the method is finally demonstrated for a selection of large acceptor molecules.
中文翻译:

通过线性回归将未收敛的GW能量外推到完整的基础集极限。
GW近似于电子自能是一种公认的方法,用于获得分子的电子准粒子能,尤其是其电离势和电子亲和力。尽管比相应的波函数方法要快得多,但GW能量仍然受基础完整性慢收敛的影响。此限制阻碍了GW方法的广泛应用。在这里,我们表明,仅基于具有未收敛基集的快速初步计算,就可以达到繁重的GW计算的完整基集限制。我们介绍了一个线性模型,该模型关联了104个包含H,C,O,N,F,P,S和Cl的有机分子中大约600个状态的大型数据库的分子轨道特性和基本收敛误差。该模型采用基于分子轨道的非线性描述符,该描述符有效地编码化学空间,从而提供出色的可传递性。使用少量的描述符(17),这种外推过程的性能优于早期的更多基于物理方法的方法。最终证明了该方法的预测能力可用于选择大的受体分子。
更新日期:2020-07-14
中文翻译:
通过线性回归将未收敛的GW能量外推到完整的基础集极限。
GW近似于电子自能是一种公认的方法,用于获得分子的电子准粒子能,尤其是其电离势和电子亲和力。尽管比相应的波函数方法要快得多,但GW能量仍然受基础完整性慢收敛的影响。此限制阻碍了GW方法的广泛应用。在这里,我们表明,仅基于具有未收敛基集的快速初步计算,就可以达到繁重的GW计算的完整基集限制。我们介绍了一个线性模型,该模型关联了104个包含H,C,O,N,F,P,S和Cl的有机分子中大约600个状态的大型数据库的分子轨道特性和基本收敛误差。该模型采用基于分子轨道的非线性描述符,该描述符有效地编码化学空间,从而提供出色的可传递性。使用少量的描述符(17),这种外推过程的性能优于早期的更多基于物理方法的方法。最终证明了该方法的预测能力可用于选择大的受体分子。


























 京公网安备 11010802027423号
京公网安备 11010802027423号