当前位置:
X-MOL 学术
›
Faraday Discuss.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Variational calculations of excited states via direct optimization of the orbitals in DFT.
Faraday Discussions ( IF 3.4 ) Pub Date : 2020-06-02 , DOI: 10.1039/d0fd00064g Gianluca Levi 1 , Aleksei V Ivanov , Hannes Jónsson
Faraday Discussions ( IF 3.4 ) Pub Date : 2020-06-02 , DOI: 10.1039/d0fd00064g Gianluca Levi 1 , Aleksei V Ivanov , Hannes Jónsson
Affiliation
|
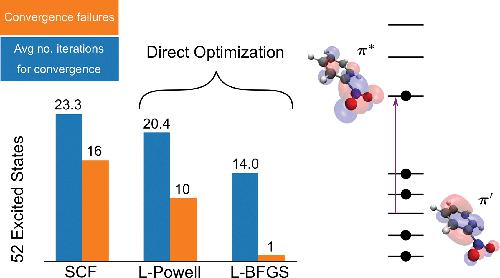
A direct optimization method for obtaining excited electronic states using density functionals is presented. It involves selective convergence on saddle points on the energy surface representing the variation of the energy as a function of the electronic degrees of freedom, thereby avoiding convergence to a minimum and corresponding variational collapse to the ground electronic state. The method is based on an exponential transformation of the molecular orbitals, making it possible to use efficient quasi-Newton optimization approaches. Direct convergence on a target nth-order saddle point is guided by an appropriate preconditioner for the optimization as well as the maximum overlap method. Results of benchmark calculations of 52 excited states of molecules indicate that the method is more robust than a standard self-consistent field (SCF) approach especially when degenerate or quasi-degenerate orbitals are involved. The method can overcome challenges arising from rearrangement of closely spaced orbitals in a charge-transfer excitation of the nitrobenzene molecule, a case where the SCF fails to converge. The formulation of the method is general and can be applied to non-unitary invariant functionals, such as self-interaction corrected functionals.
中文翻译:

通过直接优化DFT中的轨道来激发态的变分计算。
提出了一种使用密度泛函获得激发电子态的直接优化方法。它涉及在能量表面上的鞍点上进行选择性收敛,该鞍点表示能量随电子自由度的变化,从而避免收敛到最小,并避免对电子地面状态的相应变化塌陷。该方法基于分子轨道的指数变换,因此可以使用有效的拟牛顿优化方法。在目标n上直接收敛适当的预处理器可指导三阶鞍点的优化以及最大重叠法。52个分子激发态的基准计算结果表明,该方法比标准自洽场(SCF)方法更可靠,特别是在涉及简并或准简并轨道时。该方法可以克服硝基苯分子的电荷转移激发中SCF不能收敛的情况下因紧密排列的轨道重排而引起的挑战。该方法的表述是通用的,并且可以应用于非单一不变的功能,例如经自我相互作用校正的功能。
更新日期:2020-06-02
中文翻译:
通过直接优化DFT中的轨道来激发态的变分计算。
提出了一种使用密度泛函获得激发电子态的直接优化方法。它涉及在能量表面上的鞍点上进行选择性收敛,该鞍点表示能量随电子自由度的变化,从而避免收敛到最小,并避免对电子地面状态的相应变化塌陷。该方法基于分子轨道的指数变换,因此可以使用有效的拟牛顿优化方法。在目标n上直接收敛适当的预处理器可指导三阶鞍点的优化以及最大重叠法。52个分子激发态的基准计算结果表明,该方法比标准自洽场(SCF)方法更可靠,特别是在涉及简并或准简并轨道时。该方法可以克服硝基苯分子的电荷转移激发中SCF不能收敛的情况下因紧密排列的轨道重排而引起的挑战。该方法的表述是通用的,并且可以应用于非单一不变的功能,例如经自我相互作用校正的功能。

























 京公网安备 11010802027423号
京公网安备 11010802027423号