当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Accelerating Ab Initio Simulation via Nested Monte Carlo and Machine Learned Reference Potentials.
The Journal of Physical Chemistry B ( IF 3.3 ) Pub Date : 2020-06-02 , DOI: 10.1021/acs.jpcb.0c03738 Ryan B Jadrich 1, 2 , Jeffery A Leiding 1
The Journal of Physical Chemistry B ( IF 3.3 ) Pub Date : 2020-06-02 , DOI: 10.1021/acs.jpcb.0c03738 Ryan B Jadrich 1, 2 , Jeffery A Leiding 1
Affiliation
|
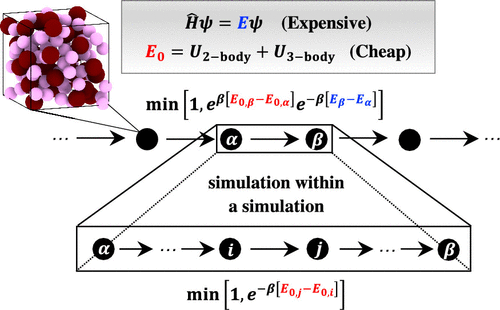
As a corollary of the rapid advances in computing, ab initio simulation is playing an increasingly important role in modeling materials at the atomic scale. Two strategies are possible, ab initio Monte Carlo (AIMC) and molecular dynamics (AIMD) simulation. The former benefits from exact sampling from the correct thermodynamic distribution, while the latter is typically more efficient with its collective all-atom coordinate updates. Here, using a relatively simple test model comprised of helium and argon, we show that AIMC can be brought up to, and even above, the performance levels of AIMD via a hybrid nested sampling/machine learning (ML) strategy. Here, ML provides an accurate classical reference potential (up to three-body explicit interactions) that can pilot long collective Monte Carlo moves that are accepted or rejected in toto à la nested Monte Carlo (NMC); this is in contrast to the single move nature of a naive implementation. Our proposed method only requires a small up front expense from evaluating the ab initio energies and forces of  (100) random configurations for training. Importantly, our method does not totally rely on the trained, assuredly imperfect, interaction. We show that high performance and exact sampling at the desired level of theory can be realized even when the trained interaction has appreciable differences from the ab initio potential. Remarkably, at the highest levels of performance realized via our approach, a pair of statistically uncorrelated atomic configurations can be generated with
(100) random configurations for training. Importantly, our method does not totally rely on the trained, assuredly imperfect, interaction. We show that high performance and exact sampling at the desired level of theory can be realized even when the trained interaction has appreciable differences from the ab initio potential. Remarkably, at the highest levels of performance realized via our approach, a pair of statistically uncorrelated atomic configurations can be generated with  (1) ab initio calculations.
(1) ab initio calculations.
中文翻译:

通过嵌套的蒙特卡洛(Monte Carlo)和机器学习的参考电位来加速从头计算。
作为计算快速发展的必然结果,从头开始仿真在原子级材料建模中扮演着越来越重要的角色。从头开始有两种策略蒙特卡洛(AIMC)和分子动力学(AIMD)模拟。前者得益于来自正确热力学分布的精确采样,而后者通常具有集体全原子坐标更新的更高效率。在这里,使用由氦和氩组成的相对简单的测试模型,我们表明,通过混合嵌套采样/机器学习(ML)策略,可以使AIMC达到甚至超过AIMD的性能水平。在这里,ML提供了准确的经典参考电位(最多三体显式交互作用),可以引导被接受或拒绝的长时间集体蒙特卡洛运动。嵌套蒙特卡洛(NMC);这与天真的实现的单动特性相反。我们提出的方法只需要很小的前期费用即可从评估训练的(100)随机配置的从头开始能量和力。重要的是,我们的方法并不完全依赖于经过训练的,肯定不完美的交互。我们表明,即使训练有素的互动与从头算的潜力有明显的差异,也可以在所需的理论水平上实现高性能和精确的采样。值得注意的是,通过我们的方法实现的最高性能水平,可以通过(1)从头算起生成一对统计上不相关的原子构型。
更新日期:2020-07-02
(100) random configurations for training. Importantly, our method does not totally rely on the trained, assuredly imperfect, interaction. We show that high performance and exact sampling at the desired level of theory can be realized even when the trained interaction has appreciable differences from the ab initio potential. Remarkably, at the highest levels of performance realized via our approach, a pair of statistically uncorrelated atomic configurations can be generated with (1) ab initio calculations.
中文翻译:
通过嵌套的蒙特卡洛(Monte Carlo)和机器学习的参考电位来加速从头计算。
作为计算快速发展的必然结果,从头开始仿真在原子级材料建模中扮演着越来越重要的角色。从头开始有两种策略蒙特卡洛(AIMC)和分子动力学(AIMD)模拟。前者得益于来自正确热力学分布的精确采样,而后者通常具有集体全原子坐标更新的更高效率。在这里,使用由氦和氩组成的相对简单的测试模型,我们表明,通过混合嵌套采样/机器学习(ML)策略,可以使AIMC达到甚至超过AIMD的性能水平。在这里,ML提供了准确的经典参考电位(最多三体显式交互作用),可以引导被接受或拒绝的长时间集体蒙特卡洛运动。嵌套蒙特卡洛(NMC);这与天真的实现的单动特性相反。我们提出的方法只需要很小的前期费用即可从评估训练的(100)随机配置的从头开始能量和力
。重要的是,我们的方法并不完全依赖于经过训练的,肯定不完美的交互。我们表明,即使训练有素的互动与从头算的潜力有明显的差异,也可以在所需的理论水平上实现高性能和精确的采样。值得注意的是,通过我们的方法实现的最高性能水平,可以通过(1)从头算起生成一对统计上不相关的原子构型。

























 京公网安备 11010802027423号
京公网安备 11010802027423号