当前位置:
X-MOL 学术
›
J. Mol. Struct.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Improved bond length determination technique for C3 and other linear molecules with a large amplitude bending vibration
Journal of Molecular Structure ( IF 3.8 ) Pub Date : 2020-11-01 , DOI: 10.1016/j.molstruc.2020.128329 Alexander A. Breier , Thomas F. Giesen , Stephen C. Ross , Koichi M.T. Yamada
Journal of Molecular Structure ( IF 3.8 ) Pub Date : 2020-11-01 , DOI: 10.1016/j.molstruc.2020.128329 Alexander A. Breier , Thomas F. Giesen , Stephen C. Ross , Koichi M.T. Yamada
|
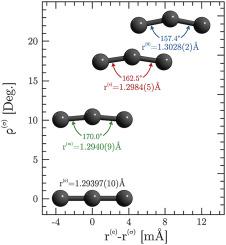
Abstract A variety of standard techniques exist for estimating molecular bond lengths from spectroscopic data of the vibrational ground state. In typical order of increasing accuracy these different estimates are known as r 0 , r s , and r m . However, for the C3 molecule each of these spectroscopically determined values is very different from that obtained in a recent high-quality ab initio calculation. We ascribe this difference to the large-amplitude ν 2 bending vibration of C3. By recognizing that vibrational averaging means that the molecule is effectively bent, even in the ground vibrational state, we develop a simple new method for estimating the bond length. This involves approximating the rotational parameter along the axis that becomes the molecular axis at linearity as A 0 = ν 2 2 . This value is then used in the evaluation of r m , rather than assuming the molecule is linear. We test this approximation for A 0 and also show that the C–C bond length we obtain is in better agreement, by an order of magnitude, with the ab initio value than any of the standard linear approaches.
中文翻译:

改进的 C3 和其他具有大幅度弯曲振动的线性分子的键长测定技术
摘要 有多种标准技术可用于根据振动基态的光谱数据估算分子键长。在提高准确性的典型顺序中,这些不同的估计被称为 r 0 、 rs 和 rm 。然而,对于 C3 分子,这些光谱确定的值中的每一个都与最近的高质量 ab initio 计算中获得的值大不相同。我们将这种差异归因于 C3 的大振幅 ν 2 弯曲振动。通过认识到振动平均意味着分子有效弯曲,即使在基振动状态下,我们开发了一种简单的新方法来估计键长。这涉及将沿线性成为分子轴的轴的旋转参数近似为 A 0 = ν 2 2 。然后将该值用于评估 rm ,而不是假设分子是线性的。我们测试了 A 0 的这个近似值,并且还表明我们获得的 C-C 键长与 ab initio 值比任何标准线性方法更符合一个数量级。
更新日期:2020-11-01
中文翻译:
改进的 C3 和其他具有大幅度弯曲振动的线性分子的键长测定技术
摘要 有多种标准技术可用于根据振动基态的光谱数据估算分子键长。在提高准确性的典型顺序中,这些不同的估计被称为 r 0 、 rs 和 rm 。然而,对于 C3 分子,这些光谱确定的值中的每一个都与最近的高质量 ab initio 计算中获得的值大不相同。我们将这种差异归因于 C3 的大振幅 ν 2 弯曲振动。通过认识到振动平均意味着分子有效弯曲,即使在基振动状态下,我们开发了一种简单的新方法来估计键长。这涉及将沿线性成为分子轴的轴的旋转参数近似为 A 0 = ν 2 2 。然后将该值用于评估 rm ,而不是假设分子是线性的。我们测试了 A 0 的这个近似值,并且还表明我们获得的 C-C 键长与 ab initio 值比任何标准线性方法更符合一个数量级。


























 京公网安备 11010802027423号
京公网安备 11010802027423号