Journal of Molecular and Cellular Cardiology ( IF 5 ) Pub Date : 2020-05-30 , DOI: 10.1016/j.yjmcc.2020.05.018 Tao He 1 , Jiale Huang 2 , Lan Chen 2 , Gang Han 3 , David Stanmore 2 , Jutta Krebs-Haupenthal 2 , Metin Avkiran 4 , Marco Hagenmüller 2 , Johannes Backs 2
|
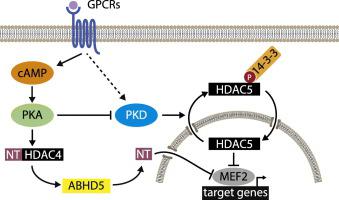
Class IIa histone deacetylases (HDACs) critically regulate cardiac function through the repression of the activity of myocyte enhancer factor 2 (MEF2)-dependent gene programs. Protein kinase D (PKD) and Ca2+/Calmodulin-dependent kinase II (CaMKII) activate MEF2 by phosphorylating distinct HDAC isoforms and thereby creating 14–3-3 binding sites for nucleo-cytoplasmic shuttling. Recently, it has been shown that this process is counteracted by cyclic AMP (cAMP)-dependent signaling. Here, we investigated the specific mechanisms of how cAMP-dependent signaling regulates distinct HDAC isoforms and determined their relative contributions to the protection from pathological MEF2 activation. We found that cAMP is sufficient to induce nuclear retention and to blunt phosphorylation of the 14–3-3 binding sites of HDAC5 (Ser259/498) and HDAC9 (Ser218/448) but not HDAC4 (Ser246/467/632). These regulatory events could be observed only in cardiomyocytes and myocyte-like cells but not in non-myocytes, pointing to an indirect myocyte-specific mode of action. Consistent with one previous report, we found that blunted phosphorylation of HDAC5 and HDAC9 was mediated by protein kinase A (PKA)-dependent inhibition of PKD. However, we show by the use of neonatal cardiomyocytes derived from genetic HDAC mouse models that endogenous HDAC5 but not HDAC9 contributes specifically to the repression of endogenous MEF2 activity. HDAC4 contributed significantly to the repression of MEF2 activity but based on the mechanistic findings of this study combined with previous results we attribute this to PKA-dependent proteolysis of HDAC4. Consistently, cAMP-induced repression of agonist-driven cellular hypertrophy was blunted in cardiomyocytes deficient for both HDAC5 and HDAC4. In conclusion, cAMP inhibits MEF2 through both nuclear accumulation of hypo-phosphorylated HDAC5 and through a distinct HDAC4-dependent mechanism.
中文翻译:
环 AMP 通过 HDAC5 的肌细胞特异性低磷酸化抑制病理性 MEF2 激活。
IIa 类组蛋白去乙酰化酶 (HDAC) 通过抑制肌细胞增强因子 2 (MEF2) 依赖性基因程序的活性来严格调节心脏功能。蛋白激酶 D (PKD) 和 Ca 2+/钙调蛋白依赖性激酶 II (CaMKII) 通过磷酸化不同的 HDAC 异构体来激活 MEF2,从而为核质穿梭创造 14-3-3 个结合位点。最近,已经表明该过程被依赖于环 AMP (cAMP) 的信号所抵消。在这里,我们研究了 cAMP 依赖性信号如何调节不同 HDAC 同种型的具体机制,并确定了它们对保护免于病理性 MEF2 激活的相对贡献。我们发现 cAMP 足以诱导核滞留并减弱 HDAC5 (Ser259/498) 和 HDAC9 (Ser218/448) 的 14-3-3 结合位点的磷酸化,但不能抑制 HDAC4 (Ser246/467/632)。这些调节事件只能在心肌细胞和肌细胞样细胞中观察到,而不能在非肌细胞中观察到,表明间接的心肌细胞特异性作用模式。与之前的一份报告一致,我们发现 HDAC5 和 HDAC9 的钝磷酸化是由蛋白激酶 A (PKA) 依赖的 PKD 抑制介导的。然而,我们通过使用源自遗传 HDAC 小鼠模型的新生儿心肌细胞表明,内源性 HDAC5 而不是 HDAC9,特别有助于抑制内源性 MEF2 活性。HDAC4 对 MEF2 活性的抑制做出了显着贡献,但根据本研究的机制发现和先前的结果,我们将其归因于 HDAC4 的 PKA 依赖性蛋白水解。一致地,在缺乏 HDAC5 和 HDAC4 的心肌细胞中,cAMP 诱导的对激动剂驱动的细胞肥大的抑制减弱。综上所述,


























 京公网安备 11010802027423号
京公网安备 11010802027423号