当前位置:
X-MOL 学术
›
Int. J. Quantum Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Embedded, graph-theoretically defined many-body approximations for wavefunction-in-DFT and DFT-in-DFT: Applications to gas- and condensed-phase ab initio molecular dynamics, and potential surfaces for quantum nuclear effects
International Journal of Quantum Chemistry ( IF 2.2 ) Pub Date : 2020-05-26 , DOI: 10.1002/qua.26244 Timothy C. Ricard 1 , Anup Kumar 1 , Srinivasan S. Iyengar 1
International Journal of Quantum Chemistry ( IF 2.2 ) Pub Date : 2020-05-26 , DOI: 10.1002/qua.26244 Timothy C. Ricard 1 , Anup Kumar 1 , Srinivasan S. Iyengar 1
Affiliation
|
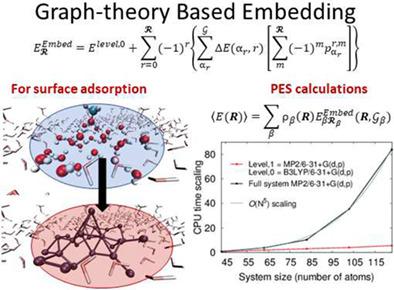
Funding information XSEDE, Grant/Award Number: CHE170081; National Science Foundation, Grant/Award Number: CHE-1665336 Abstract We present a graph-theoretic approach to adaptively compute many-body approximations in an efficient manner to perform (a) accurate post-Hartree–Fock (HF) ab initio molecular dynamics (AIMD) at density functional theory (DFT) cost for mediumto large-sized molecular clusters, (b) hybrid DFT electronic structure calculations for condensed-phase simulations at the cost of pure density functionals, (c) reduced-cost on-the-fly basis extrapolation for gas-phase AIMD and condensed phase studies, and (d) accurate post-HF-level potential energy surfaces at DFT cost for quantum nuclear effects. The salient features of our approach are ONIOM-like in that (a) the full system (cluster or condensed phase) calculation is performed at a lower level of theory (pure DFT for condensed phase or hybrid DFT for molecular systems), and (b) this approximation is improved through a correction term that captures all many-body interactions up to any given order within a higher level of theory (hybrid DFT for condensed phase; CCSD or MP2 for cluster), combined through graph-theoretic methods. Specifically, a region of chemical interest is coarse-grained into a set of nodes and these nodes are then connected to form edges based on a given definition of local envelope (or threshold) of interactions. The nodes and edges together define a graph, which forms the basis for developing the many-body expansion. The methods are demonstrated through (a) ab initio dynamics studies on protonated water clusters and polypeptide fragments, (b) potential energy surface calculations on one-dimensional water chains such as those found in ion channels, and (c) conformational stabilization and lattice energy studies on homogeneous and heterogeneous surfaces of water with organic adsorbates using two-dimensional periodic boundary conditions.
中文翻译:

嵌入式、图形理论定义的 DFT 波函数和 DFT-in-DFT 的多体近似:在气相和凝聚相 ab initio 分子动力学中的应用,以及量子核效应的潜在表面
资金信息 XSEDE,资助/奖励编号:CHE170081;美国国家科学基金会,资助/奖项编号:CHE-1665336 摘要我们提出了一种图论方法,以有效的方式自适应地计算多体近似值,以执行 (a) 准确的后 Hartree-Fock (HF) ab initio 分子动力学( AIMD) 以密度泛函理论 (DFT) 为中到大型分子簇的成本,(b) 以纯密度泛函为代价的凝聚相模拟的混合 DFT 电子结构计算,(c) 即时降低成本用于气相 AIMD 和凝聚相研究的基础外推,以及 (d) 以 DFT 为代价的精确后 HF 级势能面,用于量子核效应。我们的方法的显着特征类似于 ONIOM,因为 (a) 整个系统(簇或凝聚相)计算是在较低的理论水平(凝聚相的纯 DFT 或分子系统的混合 DFT)进行的,和 (b ) 这种近似通过一个校正项得到改进,该校正项在更高的理论水平(凝聚相的混合 DFT;簇的 CCSD 或 MP2)中捕获所有多体相互作用,直到任何给定的阶数,并通过图论方法组合。具体而言,将感兴趣的化学区域粗粒度化为一组节点,然后将这些节点连接起来,根据给定的相互作用的局部包络(或阈值)定义形成边。节点和边一起定义了一个图,它构成了开发多体扩展的基础。
更新日期:2020-05-26
中文翻译:
嵌入式、图形理论定义的 DFT 波函数和 DFT-in-DFT 的多体近似:在气相和凝聚相 ab initio 分子动力学中的应用,以及量子核效应的潜在表面
资金信息 XSEDE,资助/奖励编号:CHE170081;美国国家科学基金会,资助/奖项编号:CHE-1665336 摘要我们提出了一种图论方法,以有效的方式自适应地计算多体近似值,以执行 (a) 准确的后 Hartree-Fock (HF) ab initio 分子动力学( AIMD) 以密度泛函理论 (DFT) 为中到大型分子簇的成本,(b) 以纯密度泛函为代价的凝聚相模拟的混合 DFT 电子结构计算,(c) 即时降低成本用于气相 AIMD 和凝聚相研究的基础外推,以及 (d) 以 DFT 为代价的精确后 HF 级势能面,用于量子核效应。我们的方法的显着特征类似于 ONIOM,因为 (a) 整个系统(簇或凝聚相)计算是在较低的理论水平(凝聚相的纯 DFT 或分子系统的混合 DFT)进行的,和 (b ) 这种近似通过一个校正项得到改进,该校正项在更高的理论水平(凝聚相的混合 DFT;簇的 CCSD 或 MP2)中捕获所有多体相互作用,直到任何给定的阶数,并通过图论方法组合。具体而言,将感兴趣的化学区域粗粒度化为一组节点,然后将这些节点连接起来,根据给定的相互作用的局部包络(或阈值)定义形成边。节点和边一起定义了一个图,它构成了开发多体扩展的基础。


























 京公网安备 11010802027423号
京公网安备 11010802027423号