当前位置:
X-MOL 学术
›
J. Mol. Struct.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
XRD structure and vibrational analysis of DL-β-Leucine, as aided by DFT tetramer model and characterized by NBO, AIM and NCI calculations
Journal of Molecular Structure ( IF 3.8 ) Pub Date : 2020-10-01 , DOI: 10.1016/j.molstruc.2020.128495 Madhuri D. Prabhu , J. Tonannavar Yenagi , Vinayak Kamat , J. Tonannavar
Journal of Molecular Structure ( IF 3.8 ) Pub Date : 2020-10-01 , DOI: 10.1016/j.molstruc.2020.128495 Madhuri D. Prabhu , J. Tonannavar Yenagi , Vinayak Kamat , J. Tonannavar
|
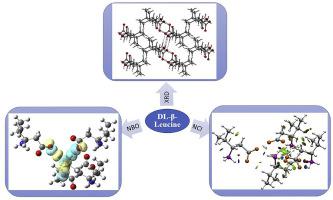
Abstract Vibrational spectra due to molecular vibrations of amino acids are influenced by their zwitterionicity and oligomerization as defined by H-bonds. When these two influencing factors are incorporated into computational modeling, a very good agreement between theory and experiment is achieved. In the present work we show this good agreement for the unnatural amino acid, DL-β-Leucine. From a single-crystal X-ray diffraction analysis, DL-β-Leucine has been found to be a zwitterion in the triclinic space group P-1, having three distinct N–H⋯O bonds from a cationic –NH3+ and anionic –CO2‒ moieties. This structure has been utilized as a model for building a tetramer at B3LYP/6-311G (d,p) level. It is shown that each of the three N–H bonds in the –NH3+ of a central S-enantiomer in the tetramer is H-bonded with the oxygen atoms in the –CO2‒ moiety belonging to its three neighbour molecules. Experimentally rich vibrational spectra of IR and Raman bands, especially belonging to the NH3+ and –CO2‒ moieties agree very well with the tetramer modes, including Raman bands below 200 cm−1 generically called ‘lattice modes’. As for the electronic characterization of N–H⋯O bonds, AIM calculation gives bond energies of −6.44, −9.67 and −11.24 kcal/mol at bond critical points (BCP: 3, −1), with relative changes that are consistent with the geometrical parameters as well as IR spectral shifts of the N–H⋯O bonds. This is further supported by NBO analysis as well. Further characterization of N–H⋯O bonds as noncovalent interaction by reduced density gradient analysis in relation to van der Waals and steric interactions has been shown to be consistent with AIM and NBO results.
中文翻译:

DL-β-亮氨酸的 XRD 结构和振动分析,在 DFT 四聚体模型的辅助下,并通过 NBO、AIM 和 NCI 计算表征
摘要 氨基酸分子振动引起的振动光谱受其两性离子性和由氢键定义的寡聚化的影响。当这两个影响因素被纳入计算建模时,理论和实验之间达到了很好的一致性。在目前的工作中,我们对非天然氨基酸 DL-β-亮氨酸显示了这种良好的一致性。从单晶 X 射线衍射分析中,已发现 DL-β-亮氨酸是三斜空间群 P-1 中的两性离子,具有来自阳离子 -NH3+ 和阴离子 -CO2 的三个不同的 N-H⋯O 键- 部分。这种结构已被用作构建 B3LYP/6-311G (d,p) 水平四聚体的模型。结果表明,四聚体中心 S-对映体的 –NH3+ 中的三个 N–H 键中的每一个都与属于其三个相邻分子的 –CO2– 部分中的氧原子 H 键合。IR 和拉曼带的实验丰富的振动光谱,特别是属于 NH3+ 和 -CO2- 部分,与四聚体模式非常吻合,包括低于 200 cm-1 的拉曼带,通常称为“晶格模式”。对于 N–H⋯O 键的电子表征,AIM 计算得出键能在键临界点 (BCP: 3, -1) 分别为 -6.44、-9.67 和 -11.24 kcal/mol,相对变化与N-H⋯O 键的几何参数以及红外光谱位移。这也得到了 NBO 分析的进一步支持。
更新日期:2020-10-01
中文翻译:
DL-β-亮氨酸的 XRD 结构和振动分析,在 DFT 四聚体模型的辅助下,并通过 NBO、AIM 和 NCI 计算表征
摘要 氨基酸分子振动引起的振动光谱受其两性离子性和由氢键定义的寡聚化的影响。当这两个影响因素被纳入计算建模时,理论和实验之间达到了很好的一致性。在目前的工作中,我们对非天然氨基酸 DL-β-亮氨酸显示了这种良好的一致性。从单晶 X 射线衍射分析中,已发现 DL-β-亮氨酸是三斜空间群 P-1 中的两性离子,具有来自阳离子 -NH3+ 和阴离子 -CO2 的三个不同的 N-H⋯O 键- 部分。这种结构已被用作构建 B3LYP/6-311G (d,p) 水平四聚体的模型。结果表明,四聚体中心 S-对映体的 –NH3+ 中的三个 N–H 键中的每一个都与属于其三个相邻分子的 –CO2– 部分中的氧原子 H 键合。IR 和拉曼带的实验丰富的振动光谱,特别是属于 NH3+ 和 -CO2- 部分,与四聚体模式非常吻合,包括低于 200 cm-1 的拉曼带,通常称为“晶格模式”。对于 N–H⋯O 键的电子表征,AIM 计算得出键能在键临界点 (BCP: 3, -1) 分别为 -6.44、-9.67 和 -11.24 kcal/mol,相对变化与N-H⋯O 键的几何参数以及红外光谱位移。这也得到了 NBO 分析的进一步支持。

























 京公网安备 11010802027423号
京公网安备 11010802027423号