当前位置:
X-MOL 学术
›
Macromol. Theor. Simul.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Conformation and Diffusivity of Ring and Linear Polyethylene Oxide in Aqueous Solution: Molecular Topology Dependent Concentration Effects and Comparison with Experimental Data
Macromolecular Theory and Simulations ( IF 1.4 ) Pub Date : 2020-05-19 , DOI: 10.1002/mats.202000016 Dimitrios G. Tsalikis 1 , Terpsichori S. Alexiou 1 , Panagiotis V. Alatas 1 , Vlasis G. Mavrantzas 1, 2
Macromolecular Theory and Simulations ( IF 1.4 ) Pub Date : 2020-05-19 , DOI: 10.1002/mats.202000016 Dimitrios G. Tsalikis 1 , Terpsichori S. Alexiou 1 , Panagiotis V. Alatas 1 , Vlasis G. Mavrantzas 1, 2
Affiliation
|
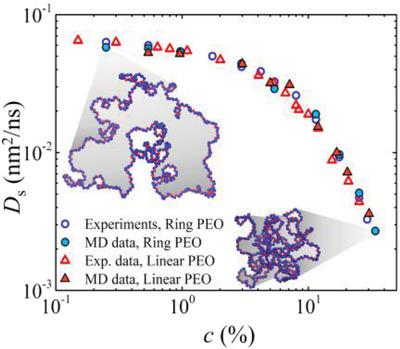
Atomistic molecular dynamics simulations are performed on aqueous solutions of ring and linear polyethylene oxide (PEO) under good solvent conditions for the investigation of their conformation and diffusion dynamics. The systems simulated span a wide range of molecular weights (from 2 to 10 k) and polymer concentrations. In the limit of infinite dilution, both conformational and dynamic properties appear to be practically molecular topology independent as far as their dependence on chain length and concentration is concerned, and a very favorable agreement is observed with respect to relevant well‐established theoretical predictions, experimental data, and earlier simulation studies. Crossing over to the semidilute regime, molecular topology starts to bear a distinct effect on the observed conformations of the two different molecular geometries. On the other hand, the diffusion dynamics of PEO in the semidilute regime appears to be practically molecular topology independent judging from the dependence of the chain center‐of‐mass diffusivity on chain length and concentration. Indeed, and despite the fact that ring molecules exhibit consistently a faster dynamics than their linear homologs over the entire concentration range simulated, the estimated concentration scalings for both types of architecture are strikingly similar. The findings of this work are compared with scaling arguments and available experimental data.
中文翻译:

环和线性聚环氧乙烷在水溶液中的构象和扩散率:分子拓扑学相关的浓度效应并与实验数据进行比较
在良好的溶剂条件下,对环和线性聚环氧乙烷(PEO)的水溶液进行原子分子动力学模拟,以研究其构象和扩散动力学。模拟的系统涵盖了分子量(2至10 k)和聚合物浓度的广泛范围。在无限稀释的范围内,就其对链长和浓度的依赖性而言,构象和动力学性质似乎都是分子拓扑学上独立的,并且在相关的既定理论预测,实验方面观察到非常有利的协议。数据和较早的模拟研究。越过半稀释状态,分子拓扑开始对观察到的两种不同分子几何构型产生明显影响。另一方面,从链的质心扩散率对链长和浓度的依赖性来看,半稀释状态下PEO的扩散动力学似乎与分子拓扑学无关。确实,尽管事实上在模拟的整个浓度范围内,环分子始终比其线性同系物表现出更快的动力学,但两种结构类型的估计浓度比例却惊人地相似。将这项工作的发现与缩放参数和可用的实验数据进行比较。尽管在模拟的整个浓度范围内,环分子始终比线性同系物表现出更快的动力学,但两种结构类型的估计浓度比例却极为相似。将这项工作的发现与缩放参数和可用的实验数据进行比较。尽管在模拟的整个浓度范围内,环分子始终比线性同系物表现出更快的动力学,但两种结构类型的估计浓度比例却极为相似。将这项工作的发现与缩放参数和可用的实验数据进行比较。
更新日期:2020-05-19
中文翻译:
环和线性聚环氧乙烷在水溶液中的构象和扩散率:分子拓扑学相关的浓度效应并与实验数据进行比较
在良好的溶剂条件下,对环和线性聚环氧乙烷(PEO)的水溶液进行原子分子动力学模拟,以研究其构象和扩散动力学。模拟的系统涵盖了分子量(2至10 k)和聚合物浓度的广泛范围。在无限稀释的范围内,就其对链长和浓度的依赖性而言,构象和动力学性质似乎都是分子拓扑学上独立的,并且在相关的既定理论预测,实验方面观察到非常有利的协议。数据和较早的模拟研究。越过半稀释状态,分子拓扑开始对观察到的两种不同分子几何构型产生明显影响。另一方面,从链的质心扩散率对链长和浓度的依赖性来看,半稀释状态下PEO的扩散动力学似乎与分子拓扑学无关。确实,尽管事实上在模拟的整个浓度范围内,环分子始终比其线性同系物表现出更快的动力学,但两种结构类型的估计浓度比例却惊人地相似。将这项工作的发现与缩放参数和可用的实验数据进行比较。尽管在模拟的整个浓度范围内,环分子始终比线性同系物表现出更快的动力学,但两种结构类型的估计浓度比例却极为相似。将这项工作的发现与缩放参数和可用的实验数据进行比较。尽管在模拟的整个浓度范围内,环分子始终比线性同系物表现出更快的动力学,但两种结构类型的估计浓度比例却极为相似。将这项工作的发现与缩放参数和可用的实验数据进行比较。

























 京公网安备 11010802027423号
京公网安备 11010802027423号