当前位置:
X-MOL 学术
›
ACS Cent. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery.
ACS Central Science ( IF 18.2 ) Pub Date : 2020-05-19 , DOI: 10.1021/acscentsci.0c00229 Francesco Gentile 1 , Vibudh Agrawal 1 , Michael Hsing 1 , Anh-Tien Ton 1 , Fuqiang Ban 1 , Ulf Norinder 2, 3 , Martin E Gleave 1 , Artem Cherkasov 1
ACS Central Science ( IF 18.2 ) Pub Date : 2020-05-19 , DOI: 10.1021/acscentsci.0c00229 Francesco Gentile 1 , Vibudh Agrawal 1 , Michael Hsing 1 , Anh-Tien Ton 1 , Fuqiang Ban 1 , Ulf Norinder 2, 3 , Martin E Gleave 1 , Artem Cherkasov 1
Affiliation
|
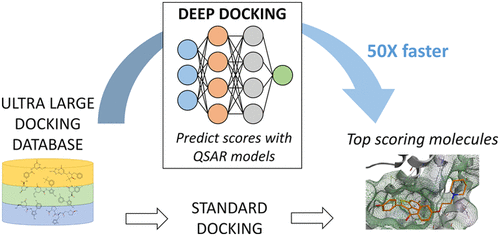
Drug discovery is a rigorous process that requires billion dollars of investments and decades of research to bring a molecule “from bench to a bedside”. While virtual docking can significantly accelerate the process of drug discovery, it ultimately lags the current rate of expansion of chemical databases that already exceed billions of molecular records. This recent surge of small molecules availability presents great drug discovery opportunities, but also demands much faster screening protocols. In order to address this challenge, we herein introduce Deep Docking (DD), a novel deep learning platform that is suitable for docking billions of molecular structures in a rapid, yet accurate fashion. The DD approach utilizes quantitative structure–activity relationship (QSAR) deep models trained on docking scores of subsets of a chemical library to approximate the docking outcome for yet unprocessed entries and, therefore, to remove unfavorable molecules in an iterative manner. The use of DD methodology in conjunction with the FRED docking program allowed rapid and accurate calculation of docking scores for 1.36 billion molecules from the ZINC15 library against 12 prominent target proteins and demonstrated up to 100-fold data reduction and 6000-fold enrichment of high scoring molecules (without notable loss of favorably docked entities). The DD protocol can readily be used in conjunction with any docking program and was made publicly available.
中文翻译:

深度扩展坞:用于增强基于结构的药物发现的深度学习平台。
药物发现是一个严格的过程,需要数十亿美元的投资和数十年的研究才能使分子“从实验台到床边”。尽管虚拟对接可以大大加快药物发现过程,但最终落后于目前已经超过数十亿分子记录的化学数据库的当前扩展速度。最近小分子可用性的激增提供了巨大的药物发现机会,但也要求更快的筛选方案。为了解决这一挑战,我们在这里介绍了Deep Docking(DD),这是一种新颖的深度学习平台,适用于以快速而准确的方式对接数十亿个分子结构。该DD该方法利用在化学文库子集的对接得分上训练的定量结构-活性关系(QSAR)深层模型来估算尚未处理的条目的对接结果,因此,以迭代的方式去除了不利的分子。使用的DD结合方法与FRED对接程序允许对接得分的快速,准确的计算用于从针对12突出的靶蛋白的ZINC15库1.36十亿分子并表现出高达高得分的100倍的数据缩减和6000倍的富集分子(没有明显损失的对接实体)。的DD协议可以很容易地与任何对接程序一起使用,并公开提供。
更新日期:2020-06-24
中文翻译:
深度扩展坞:用于增强基于结构的药物发现的深度学习平台。
药物发现是一个严格的过程,需要数十亿美元的投资和数十年的研究才能使分子“从实验台到床边”。尽管虚拟对接可以大大加快药物发现过程,但最终落后于目前已经超过数十亿分子记录的化学数据库的当前扩展速度。最近小分子可用性的激增提供了巨大的药物发现机会,但也要求更快的筛选方案。为了解决这一挑战,我们在这里介绍了Deep Docking(DD),这是一种新颖的深度学习平台,适用于以快速而准确的方式对接数十亿个分子结构。该DD该方法利用在化学文库子集的对接得分上训练的定量结构-活性关系(QSAR)深层模型来估算尚未处理的条目的对接结果,因此,以迭代的方式去除了不利的分子。使用的DD结合方法与FRED对接程序允许对接得分的快速,准确的计算用于从针对12突出的靶蛋白的ZINC15库1.36十亿分子并表现出高达高得分的100倍的数据缩减和6000倍的富集分子(没有明显损失的对接实体)。的DD协议可以很容易地与任何对接程序一起使用,并公开提供。
























 京公网安备 11010802027423号
京公网安备 11010802027423号