Computational and Theoretical Chemistry ( IF 2.8 ) Pub Date : 2020-05-11 , DOI: 10.1016/j.comptc.2020.112848 Mamona Khalid , Rasheed Ahmad Khera , Sobia Jabeen , Peter Langer , Javed Iqbal
|
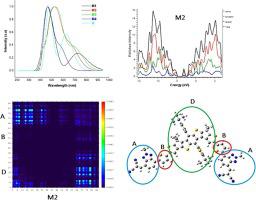
In present study, four A-D-A type of 2D conjugated molecules (M1-M4) are designed to find their opto-electronic properties. These molecules have interlinked with donor and acceptor moieties through thiophene unit, 2-(5,6-difluoro-2-methlyene-3-oxo-2,3-dihydro-1H-iden-1-ylidene)malononitrile (M1), 2-((2-methyl-2H-benzo[d][1,2,3] triazol-4-yl) methylene)malononitrile (M2), 2-((5,6-difluoro-2-methyl-2H-benzo[d][1,2,3] triazol-4-yl) methylene)malononitrile (M3), and 3-methyl-5-methylene-2-thioxothiazolidin-4-one (M4) by density functional theory (DFT). The photovoltaic properties of these newly designed molecules are evaluated and compared with reference molecule R (ITIC2) contains electron rich benzo[1,2-b:4,5-b′]di(cyclopenta[2,1-b:3,4-b′]dithiophene) core, electron-deficient end groups, conjugated 5-(2-ethylhexyl)thiophene side chains, and nonconjugated 4-hexylbenzene side chains. The effect of end acceptor groups on absorption, energy level, charge transport, morphology, and photovoltaic properties of the designed molecules (M1-M4) were investigated by TD-DFT B3LYP/6-31G basic level of theory and compared with reference molecule R. Among all molecules, M1 and M3 showed lower band gap and exhibited more absorption with B3LYP/6-31G (d, p) level of theory due to highly extended conjugation between electron withdrawing end-capped acceptor moieties. The reorganization energy calculation showed that λe of M2 and M3 was lowered because of their good charge transfer ability as compared to other molecules. M3 also showed least coefficient interaction between acceptor and donor groups in TDM analysis which showed the easier and highest dissociation at the excited state. Overall, designed structure M3 was found to be more effective and efficient acceptor molecule for solar cell application. The findings provide novel information for the development of ITIC based acceptors for OPVs.
中文翻译:
设计用于小分子有机太阳能电池的2D稠环材料
在本研究中,设计了四种ADA类型的2D共轭分子(M1 - M4),以发现它们的光电特性。这些分子通过噻吩单元2-(5,6-二氟-2-亚甲基-3-氧代-2,3-二氢-1H-亚基-1-亚烷基)丙二腈(M1)2与供体和受体部分互连-(((2-甲基-2H-苯并[d] [1,2,3]三唑-4-基)亚甲基)丙二腈(M2),2-(((5,6-二氟-2-甲基-2H-苯并通过密度泛函理论(DFT),[d] [1,2,3]三唑-4-基)亚甲基)丙二腈(M3)和3-甲基-5-亚甲基-2-硫代噻唑并恶唑烷-4-酮(M4)。对这些新设计的分子的光伏性质进行了评估,并与参考分子R进行了比较。(ITIC2)包含富电子的苯并[1,2- b:4,5- b '] di(环戊[2,1- b:3,4- b ']二噻吩)核,缺电子端基,共轭5 -(2-乙基己基)噻吩侧链和非共轭的4-己基苯侧链。通过TD-DFT B3LYP / 6-31G基本理论水平研究了末端受体基团对设计分子(M1-M4)的吸收,能级,电荷传输,形态和光伏性质的影响,并与参考分子R进行了比较。在所有分子中,M1和M3由于吸电子的末端封端的受体部分之间的高度扩展的共轭作用,在B3LYP / 6-31G(d,p)的理论水平下,Cb表现出较低的带隙并显示出更多的吸收。重组能量计算表明,λ Ë的M2和M3相比于其它分子降低的,因为它们的良好的电荷传输能力。在TDM分析中,M3还显示受体和供体基团之间的系数相互作用最小,这表明在激发态下更容易且最高解离。总体设计结构M3被发现对于太阳能电池应用是更有效的受体分子。这些发现为基于ITIC的OPV受体的开发提供了新颖的信息。


























 京公网安备 11010802027423号
京公网安备 11010802027423号