当前位置:
X-MOL 学术
›
Genes Cells
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
A simple and practical workflow for genotyping of CRISPR-Cas9-based knockout phenotypes using multiplexed amplicon sequencing.
Genes to Cells ( IF 2.1 ) Pub Date : 2020-04-23 , DOI: 10.1111/gtc.12775 Midori Iida 1 , Miyuki Suzuki 2 , Yuto Sakane 2 , Hiroyo Nishide 3 , Ikuo Uchiyama 3 , Takashi Yamamoto 2 , Ken-Ichi T Suzuki 2, 4 , Satoshi Fujii 1
Genes to Cells ( IF 2.1 ) Pub Date : 2020-04-23 , DOI: 10.1111/gtc.12775 Midori Iida 1 , Miyuki Suzuki 2 , Yuto Sakane 2 , Hiroyo Nishide 3 , Ikuo Uchiyama 3 , Takashi Yamamoto 2 , Ken-Ichi T Suzuki 2, 4 , Satoshi Fujii 1
Affiliation
|
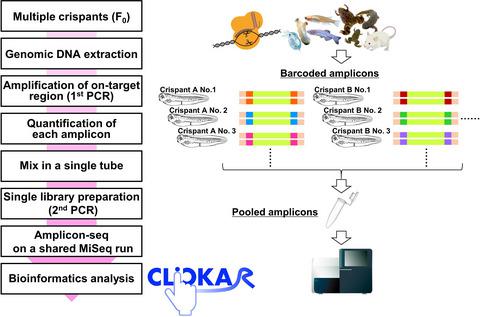
Founder animals carrying high proportions of somatic mutation induced by CRISPR–Cas9 enable a rapid and scalable strategy for the functional screening of numerous target genes in vivo. In this functional screening, genotyping using pooled amplicons with next‐generation sequencing is the most suitable approach for large‐scale management of multiple samples and accurate evaluation of the efficiency of Cas9‐induced somatic mutations at target sites. Here, we present a simple workflow for genotyping of multiple CRISPR–Cas9‐based knockout founders by pooled amplicon sequencing. Using custom barcoded primers, pooled amplicons from multiple individuals can be run in a single‐indexed library on the Illumina MiSeq platform. Additionally, a user‐friendly web tool, CLiCKAR, is available to simultaneously perform demultiplexing of pooled sequence data and evaluation of somatic mutation in each phenotype. CLiCKAR provides users with practical reports regarding the positions of insertions/deletions, as well as the frameshift ratio and tables containing mutation sequences, and read counts of each phenotype, with just a few clicks by the implementation of demultiplexing for pooled sample data and calculation of the frameshift ratio. This genotyping workflow can be harnessed to evaluate genotype–phenotype correlations in CRISPR–Cas9‐based loss‐of‐function screening of numerous target genes in various organisms.
中文翻译:

使用多重扩增子测序进行基于CRISPR-Cas9基因敲除表型基因分型的简单实用方法。
通过CRISPR–Cas9诱导的携带高比例体细胞突变的方正动物能够为体内众多靶基因的功能筛选提供快速,可扩展的策略。在此功能筛选中,使用合并的扩增子与下一代测序进行基因分型是最适合大规模管理多个样品并准确评估目标部位Cas9诱导的体细胞突变效率的方法。在这里,我们提供了一个简单的工作流程,可通过合并的扩增子测序对多个基于CRISPR–Cas9的敲除基因进行基因分型。使用定制的条形码引物,可以在Illumina MiSeq平台上的单索引库中运行来自多个个体的合并扩增子。此外,用户友好的网络工具CLiCKAR 可用于同时执行合并序列数据的多路分解和评估每种表型中的体细胞突变。CLiCKAR为用户提供有关插入/缺失位置,移码率和包含突变序列的表以及每个表型的读取计数的实用报告,只需单击几下即可实现解复用,从而汇集样本数据并计算移码率。该基因分型工作流程可用于评估基于CRISPR-Cas9的功能丧失的各种生物中的靶基因的基因型与表型的相关性。通过对合并的样本数据进行解复用并计算移码率,只需单击几下,即可读取和读取每种表型的计数。该基因分型工作流程可用于评估基于CRISPR-Cas9的功能丧失的各种生物中的靶基因的基因型与表型的相关性。通过对合并的样本数据进行解复用并计算移码率,只需单击几下,即可读取和读取每种表型的计数。该基因分型工作流程可用于评估基于CRISPR-Cas9的功能丧失的各种生物中的靶基因的基因型与表型的相关性。
更新日期:2020-04-23
中文翻译:
使用多重扩增子测序进行基于CRISPR-Cas9基因敲除表型基因分型的简单实用方法。
通过CRISPR–Cas9诱导的携带高比例体细胞突变的方正动物能够为体内众多靶基因的功能筛选提供快速,可扩展的策略。在此功能筛选中,使用合并的扩增子与下一代测序进行基因分型是最适合大规模管理多个样品并准确评估目标部位Cas9诱导的体细胞突变效率的方法。在这里,我们提供了一个简单的工作流程,可通过合并的扩增子测序对多个基于CRISPR–Cas9的敲除基因进行基因分型。使用定制的条形码引物,可以在Illumina MiSeq平台上的单索引库中运行来自多个个体的合并扩增子。此外,用户友好的网络工具CLiCKAR 可用于同时执行合并序列数据的多路分解和评估每种表型中的体细胞突变。CLiCKAR为用户提供有关插入/缺失位置,移码率和包含突变序列的表以及每个表型的读取计数的实用报告,只需单击几下即可实现解复用,从而汇集样本数据并计算移码率。该基因分型工作流程可用于评估基于CRISPR-Cas9的功能丧失的各种生物中的靶基因的基因型与表型的相关性。通过对合并的样本数据进行解复用并计算移码率,只需单击几下,即可读取和读取每种表型的计数。该基因分型工作流程可用于评估基于CRISPR-Cas9的功能丧失的各种生物中的靶基因的基因型与表型的相关性。通过对合并的样本数据进行解复用并计算移码率,只需单击几下,即可读取和读取每种表型的计数。该基因分型工作流程可用于评估基于CRISPR-Cas9的功能丧失的各种生物中的靶基因的基因型与表型的相关性。

























 京公网安备 11010802027423号
京公网安备 11010802027423号