当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Accurate prediction of relative binding affinities of a series of HIV‐1 protease inhibitors using semi‐empirical quantum mechanical charge
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2020-04-30 , DOI: 10.1002/jcc.26218 Cheng Peng 1, 2 , Jinan Wang 1 , Zhijian Xu 1, 2 , Tingting Cai 1 , Weiliang Zhu 1, 2, 3
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2020-04-30 , DOI: 10.1002/jcc.26218 Cheng Peng 1, 2 , Jinan Wang 1 , Zhijian Xu 1, 2 , Tingting Cai 1 , Weiliang Zhu 1, 2, 3
Affiliation
|
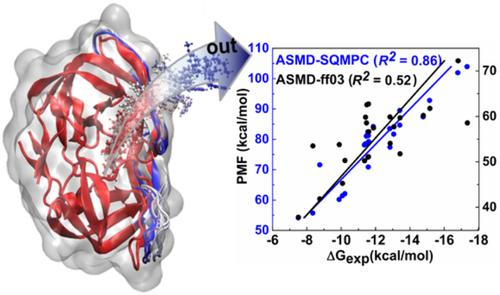
A major challenge in computer‐aided drug design is the accurate estimation of ligand binding affinity. Here, a new approach that combines the adaptive steered molecular dynamics (ASMD) and partial atomic charges calculated by semi‐empirical quantum mechanics (SQMPC), namely ASMD‐SQMPC, is suggested to predict the ligand binding affinities, with 24 HIV‐1 protease inhibitors as testing examples. In the ASMD‐SQMPC, the relative binding free energy (ΔG) is reflected by the average maximum potential of mean force (max) between bound and unbound states. The correlation coefficient (R2) between the max and experimentally determined ΔG is 0.86, showing a significant improvement compared with the conventional ASMD (R2 = 0.52). Therefore, this study provides an efficient approach to predict the relative ΔG and reveals the significance of precise partial atomic charges in the theoretical simulations.
中文翻译:

使用半经验量子力学电荷准确预测一系列 HIV-1 蛋白酶抑制剂的相对结合亲和力
计算机辅助药物设计的一个主要挑战是配体结合亲和力的准确估计。在这里,建议结合自适应导向分子动力学 (ASMD) 和半经验量子力学 (SQMPC) 计算的部分原子电荷,即 ASMD-SQMPC,来预测配体结合亲和力,与 24 HIV-1 蛋白酶抑制剂作为测试实例。在 ASMD-SQMPC 中,相对结合自由能 (ΔG) 由束缚态和未束缚态之间的平均力 (max) 的平均最大电位反映。最大值与实验确定的 ΔG 之间的相关系数 (R2) 为 0.86,与传统的 ASMD (R2 = 0.52) 相比,显示出显着改善。所以,
更新日期:2020-04-30
中文翻译:
使用半经验量子力学电荷准确预测一系列 HIV-1 蛋白酶抑制剂的相对结合亲和力
计算机辅助药物设计的一个主要挑战是配体结合亲和力的准确估计。在这里,建议结合自适应导向分子动力学 (ASMD) 和半经验量子力学 (SQMPC) 计算的部分原子电荷,即 ASMD-SQMPC,来预测配体结合亲和力,与 24 HIV-1 蛋白酶抑制剂作为测试实例。在 ASMD-SQMPC 中,相对结合自由能 (ΔG) 由束缚态和未束缚态之间的平均力 (max) 的平均最大电位反映。最大值与实验确定的 ΔG 之间的相关系数 (R2) 为 0.86,与传统的 ASMD (R2 = 0.52) 相比,显示出显着改善。所以,


























 京公网安备 11010802027423号
京公网安备 11010802027423号