Physica E: Low-dimensional Systems and Nanostructures ( IF 3.3 ) Pub Date : 2020-04-25 , DOI: 10.1016/j.physe.2020.114152 Xiaoxiao Gong , Zuoliang Ye , Shan Lu , Kuo Liu , Jiaying Liu , Zhenling Liu
|
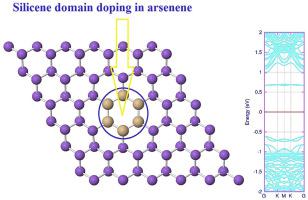
Inspired by the huge number of investigations on the applications of two-dimensional materials in nanoscale electronic devices, we examined the equilibrium structures and electronic properties of arsenene nanosheets doped with different domains of silicene, germanene, and stanene. In this regard, we replaced one hexagonal ring of arsenene with Si, Ge and Sn atoms to obtain the novel hybrid nanomaterials with unique properties. Due to the larger buckling parameter of arsenene compared to the silicene and germanene, the Si and Ge domains show less protrusion out of the surface of arsenene. On the other hand, since the Sn atom has higher atomic radius than the As atom, the protrusion observed for stanene domain in the arsenene structure will be greater. The band structure calculations show that the considered supercell of arsenene is semiconductor. The Si, Ge, and Sn domain doped monolayers also exhibit semiconductor feature, although they have narrower energy band gaps than the perfect arsenene system. Our theoretical computation would offer valuable insights into the domain doping influence on the geometric and electronic properties of the arsenene monolayer, and shed light on the application of Si-, Ge-, and Sn domain doped arsenene systems for the future nanoelectronic devices.
中文翻译:
通过锗,硅烯和锡原子团掺杂来调整砷单层的结构和电子性质
受到对二维材料在纳米级电子设备中应用的大量研究的启发,我们研究了掺有硅,锗和锡的不同区域的砷纳米片的平衡结构和电子性能。在这方面,我们用Si,Ge和Sn原子取代了一个砷的六角环,从而获得了具有独特性能的新型杂化纳米材料。由于与硅烯和锗烯相比,砷具有更大的屈曲参数,因此Si和Ge畴显示出较少的从砷表面突出的现象。另一方面,由于Sn原子具有比As原子更高的原子半径,因此在砷结构中的锡结构域观察到的突出将更大。能带结构计算表明,所考虑的砷的超级电池是半导体。Si,Ge和Sn掺杂的单层也表现出半导体特性,尽管它们的能带隙比理想的砷系统窄。我们的理论计算将为域掺杂对砷单层的几何和电子性质的影响提供有价值的见解,并为未来的纳米电子器件中Si,Ge和Sn掺杂砷系统的应用提供启示。

























 京公网安备 11010802027423号
京公网安备 11010802027423号