当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
How accurate are TD-DFT excited-state geometries compared to DFT ground-state geometries?
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2020-04-23 , DOI: 10.1002/jcc.26213 Jun Wang 1, 2 , Bo Durbeej 1
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2020-04-23 , DOI: 10.1002/jcc.26213 Jun Wang 1, 2 , Bo Durbeej 1
Affiliation
|
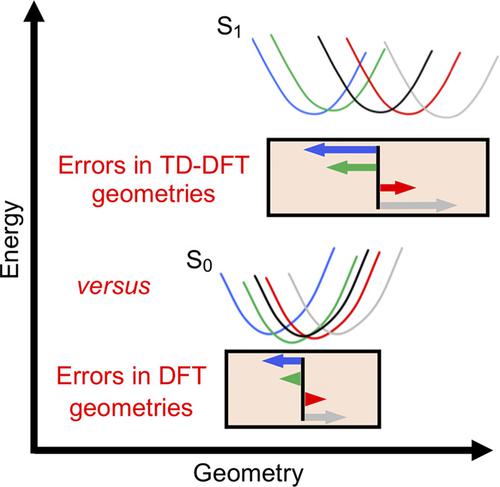
In this work, we take a different angle to the benchmarking of time‐dependent density functional theory (TD‐DFT) for the calculation of excited‐state geometries by extensively assessing how accurate such geometries are compared to ground‐state geometries calculated with ordinary DFT. To this end, we consider 20 medium‐sized aromatic organic compounds whose lowest singlet excited states are ideally suited for TD‐DFT modeling and are very well described by the approximate coupled‐cluster singles and doubles (CC2) method, and then use this method and six different density functionals (BP86, B3LYP, PBE0, M06‐2X, CAM‐B3LYP, and ωB97XD) to optimize the corresponding ground‐ and excited‐state geometries. The results show that although each hybrid functional reproduces the CC2 excited‐state bond lengths very satisfactorily, achieving an overall root mean square error of 0.011 Å for all 336 bonds in the 20 molecules, these errors are distinctly larger than those of only 0.004–0.006 Å with which the hybrid functionals reproduce the CC2 ground‐state bond lengths. Furthermore, for each functional employed, the variation in the error relative to CC2 between different molecules is found to be much larger (by at least a factor of 3) for the excited‐state geometries than for the ground‐state geometries, despite the fact that the molecules/states under investigation have rather uniform chemical and spectroscopic character. Overall, the study finds that even in favorable circumstances, TD‐DFT excited‐state geometries appear intrinsically and comparatively less accurate than DFT ground‐state ones.
中文翻译:

与 DFT 基态几何相比,TD-DFT 激发态几何的准确度如何?
在这项工作中,我们通过广泛评估激发态几何与时间相关密度泛函理论 (TD-DFT) 计算激发态几何与使用普通 DFT 计算的基态几何相比的准确程度,从不同的角度来衡量激发态几何的计算。 . 为此,我们考虑了 20 种中等大小的芳香族有机化合物,它们的最低单线激发态非常适合 TD-DFT 建模,并且可以通过近似耦合簇单双 (CC2) 方法很好地描述,然后使用这种方法和六个不同的密度泛函(BP86、B3LYP、PBE0、M06-2X、CAM-B3LYP 和 ωB97XD)来优化相应的基态和激发态几何结构。结果表明,尽管每个杂化泛函都非常令人满意地再现了 CC2 激发态键长,20 个分子中的所有 336 个键的总体均方根误差为 0.011 Å,这些误差明显大于混合泛函再现 CC2 基态键长的仅 0.004-0.006 Å 的误差。此外,对于使用的每个泛函,发现激发态几何结构的不同分子之间相对于 CC2 的误差变化比基态几何结构大得多(至少为 3 倍),尽管事实上研究中的分子/状态具有相当一致的化学和光谱特性。总体而言,该研究发现,即使在有利的情况下,TD-DFT 激发态几何结构在本质上看起来也不如 DFT 基态几何结构准确。这些误差明显大于 0.004-0.006 Å 的误差,而混合泛函用它来重现 CC2 基态键长。此外,对于使用的每个函数,发现激发态几何结构的不同分子之间相对于 CC2 的误差变化比基态几何结构大得多(至少为 3 倍),尽管事实上研究中的分子/状态具有相当一致的化学和光谱特征。总体而言,该研究发现,即使在有利的情况下,TD-DFT 激发态几何结构在本质上看起来也不如 DFT 基态几何结构准确。这些误差明显大于 0.004-0.006 Å 的误差,而混合泛函用它来重现 CC2 基态键长。此外,对于使用的每个函数,发现激发态几何结构的不同分子之间相对于 CC2 的误差变化比基态几何结构大得多(至少为 3 倍),尽管事实上研究中的分子/状态具有相当一致的化学和光谱特征。总体而言,该研究发现,即使在有利的情况下,TD-DFT 激发态几何结构在本质上看起来也不如 DFT 基态几何结构准确。发现不同分子之间相对于 CC2 的误差变化对于激发态几何比对于基态几何要大得多(至少为 3 倍),尽管事实上正在研究的分子/状态具有相当均匀的化学和光谱特性。总体而言,该研究发现,即使在有利的情况下,TD-DFT 激发态几何结构在本质上看起来也不如 DFT 基态几何结构准确。发现不同分子之间相对于 CC2 的误差变化对于激发态几何比对于基态几何要大得多(至少为 3 倍),尽管事实上正在研究的分子/状态具有相当均匀的化学和光谱特性。总体而言,该研究发现,即使在有利的情况下,TD-DFT 激发态几何结构在本质上看起来也不如 DFT 基态几何结构准确。
更新日期:2020-04-23
中文翻译:
与 DFT 基态几何相比,TD-DFT 激发态几何的准确度如何?
在这项工作中,我们通过广泛评估激发态几何与时间相关密度泛函理论 (TD-DFT) 计算激发态几何与使用普通 DFT 计算的基态几何相比的准确程度,从不同的角度来衡量激发态几何的计算。 . 为此,我们考虑了 20 种中等大小的芳香族有机化合物,它们的最低单线激发态非常适合 TD-DFT 建模,并且可以通过近似耦合簇单双 (CC2) 方法很好地描述,然后使用这种方法和六个不同的密度泛函(BP86、B3LYP、PBE0、M06-2X、CAM-B3LYP 和 ωB97XD)来优化相应的基态和激发态几何结构。结果表明,尽管每个杂化泛函都非常令人满意地再现了 CC2 激发态键长,20 个分子中的所有 336 个键的总体均方根误差为 0.011 Å,这些误差明显大于混合泛函再现 CC2 基态键长的仅 0.004-0.006 Å 的误差。此外,对于使用的每个泛函,发现激发态几何结构的不同分子之间相对于 CC2 的误差变化比基态几何结构大得多(至少为 3 倍),尽管事实上研究中的分子/状态具有相当一致的化学和光谱特性。总体而言,该研究发现,即使在有利的情况下,TD-DFT 激发态几何结构在本质上看起来也不如 DFT 基态几何结构准确。这些误差明显大于 0.004-0.006 Å 的误差,而混合泛函用它来重现 CC2 基态键长。此外,对于使用的每个函数,发现激发态几何结构的不同分子之间相对于 CC2 的误差变化比基态几何结构大得多(至少为 3 倍),尽管事实上研究中的分子/状态具有相当一致的化学和光谱特征。总体而言,该研究发现,即使在有利的情况下,TD-DFT 激发态几何结构在本质上看起来也不如 DFT 基态几何结构准确。这些误差明显大于 0.004-0.006 Å 的误差,而混合泛函用它来重现 CC2 基态键长。此外,对于使用的每个函数,发现激发态几何结构的不同分子之间相对于 CC2 的误差变化比基态几何结构大得多(至少为 3 倍),尽管事实上研究中的分子/状态具有相当一致的化学和光谱特征。总体而言,该研究发现,即使在有利的情况下,TD-DFT 激发态几何结构在本质上看起来也不如 DFT 基态几何结构准确。发现不同分子之间相对于 CC2 的误差变化对于激发态几何比对于基态几何要大得多(至少为 3 倍),尽管事实上正在研究的分子/状态具有相当均匀的化学和光谱特性。总体而言,该研究发现,即使在有利的情况下,TD-DFT 激发态几何结构在本质上看起来也不如 DFT 基态几何结构准确。发现不同分子之间相对于 CC2 的误差变化对于激发态几何比对于基态几何要大得多(至少为 3 倍),尽管事实上正在研究的分子/状态具有相当均匀的化学和光谱特性。总体而言,该研究发现,即使在有利的情况下,TD-DFT 激发态几何结构在本质上看起来也不如 DFT 基态几何结构准确。


























 京公网安备 11010802027423号
京公网安备 11010802027423号