当前位置:
X-MOL 学术
›
J. Chin. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Experimental and theoretical exploration on single crystal, structural, and quantum chemical parameters of (E)‐7‐(arylidene)‐1,2,6,7‐tetrahydro‐8H‐indeno[5,4‐b]furan‐8‐one derivatives: A comparative study
Journal of the Chinese Chemical Society ( IF 1.8 ) Pub Date : 2020-04-19 , DOI: 10.1002/jccs.202000006 Vishnu A. Adole 1, 2 , Bapu S. Jagdale 1, 2 , Thansing B. Pawar 1 , Arun B. Sawant 3
Journal of the Chinese Chemical Society ( IF 1.8 ) Pub Date : 2020-04-19 , DOI: 10.1002/jccs.202000006 Vishnu A. Adole 1, 2 , Bapu S. Jagdale 1, 2 , Thansing B. Pawar 1 , Arun B. Sawant 3
Affiliation
|
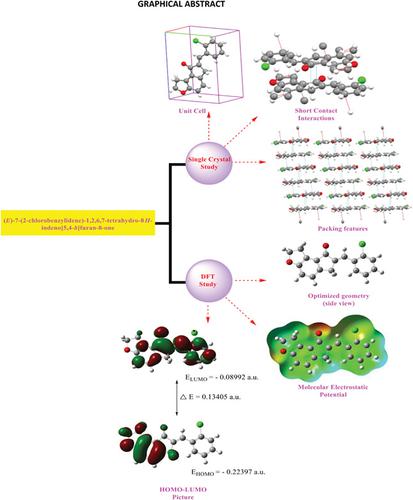
In the present research work, biologically important halogen‐substituted (E)‐7‐(arylidene)‐1,2,6,7‐tetrahydro‐8H‐indeno[5,4‐b]furan‐8‐one derivatives are studied from a structural investigation point of view. For a detailed molecular structure description, some quantum‐chemical calculations were performed by using the density functional theory method with a basis set 6–311++G(d,p). The optimized molecular geometry, bond length, atomic charges, bond angle, and harmonic vibrational frequencies have been investigated. The quantum and structural entities such as total energy, electron density distribution in highest occupied molecular orbital and lowest unoccupied molecular orbital, charge distribution, electronegativity, absolute hardness (η), softness, electrophilicity, chemical potential, and charge transfer in molecules (ΔNmax) have been computed using 6–311++G(d,p) basis set. Importantly, single‐crystal analysis study for (E)‐7‐(2‐chlorobenzylidene)‐1,2,6,7‐tetrahydro‐8H‐indeno[5,4‐b]furan‐8‐one (CBIF) has been presented. The single‐crystal examination reveals CBIF has triclinic crystal lattice with nonclassical intermolecular short contact interactions. The vibrational wavenumbers are calculated and the scaled values are compared with the experimental FT‐IR spectrum. Furthermore, the molecular electrostatic potential (MEP) and some thermodynamic functions were also explored using computational work. The theoretical geometrical parameters have been compared with the experimental results obtained from the single‐crystal examination.
中文翻译:

(E)-7-(亚芳基)-1,2,6,7-四氢-8H-茚并[5,4-b]呋喃-8-one的单晶,结构和量子化学参数的实验和理论探索衍生工具:一项比较研究
在当前的研究工作中,对具有生物学重要意义的卤素取代的(E)-7(芳基)-1,2,6,7-四氢-8 H-茚并[5,4 - b ]呋喃-8-一衍生物进行了研究从结构调查的角度来看。为了进行详细的分子结构描述,使用密度泛函理论方法以6-311 ++ G(d,p)为基础进行了一些量子化学计算。研究了优化的分子几何结构,键长,原子电荷,键角和谐波振动频率。量子和结构实体,例如总能量,在最高占据分子轨道和最低未占据分子轨道中的电子密度分布,电荷分布,电负性,绝对硬度(η),柔软性,亲电性,化学势和电荷在分子转移(Δ Ñ最大值)已经使用6-311 ++ G(d,p)基组来计算。重要的是,对(E)-7(2-氯苄叉)-1,2,6,7-四氢-8 H-茚三酮[5,4- b]进行单晶分析研究已经提出了呋喃-8-1(CBIF)。单晶检查表明,CBIF具有三斜晶格,具有非经典的分子间短接触相互作用。计算振动波数,并将标定值与实验FT-IR谱进行比较。此外,还利用计算工作探索了分子静电势(MEP)和一些热力学函数。理论几何参数已与单晶检查获得的实验结果进行了比较。
更新日期:2020-04-19
中文翻译:
(E)-7-(亚芳基)-1,2,6,7-四氢-8H-茚并[5,4-b]呋喃-8-one的单晶,结构和量子化学参数的实验和理论探索衍生工具:一项比较研究
在当前的研究工作中,对具有生物学重要意义的卤素取代的(E)-7(芳基)-1,2,6,7-四氢-8 H-茚并[5,4 - b ]呋喃-8-一衍生物进行了研究从结构调查的角度来看。为了进行详细的分子结构描述,使用密度泛函理论方法以6-311 ++ G(d,p)为基础进行了一些量子化学计算。研究了优化的分子几何结构,键长,原子电荷,键角和谐波振动频率。量子和结构实体,例如总能量,在最高占据分子轨道和最低未占据分子轨道中的电子密度分布,电荷分布,电负性,绝对硬度(η),柔软性,亲电性,化学势和电荷在分子转移(Δ Ñ最大值)已经使用6-311 ++ G(d,p)基组来计算。重要的是,对(E)-7(2-氯苄叉)-1,2,6,7-四氢-8 H-茚三酮[5,4- b]进行单晶分析研究已经提出了呋喃-8-1(CBIF)。单晶检查表明,CBIF具有三斜晶格,具有非经典的分子间短接触相互作用。计算振动波数,并将标定值与实验FT-IR谱进行比较。此外,还利用计算工作探索了分子静电势(MEP)和一些热力学函数。理论几何参数已与单晶检查获得的实验结果进行了比较。


























 京公网安备 11010802027423号
京公网安备 11010802027423号