当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
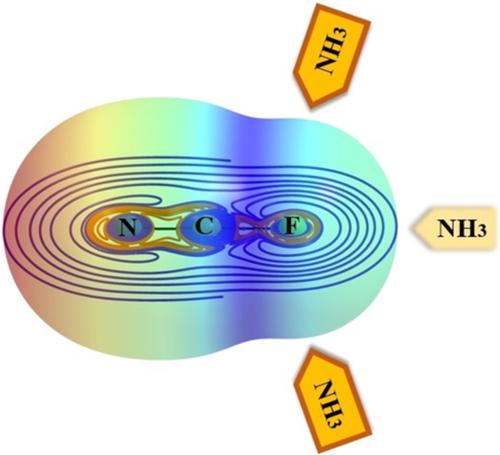
Fluorine as a Lewis acid: A symmetry‐adapted perturbation theory based on density functional theory and interacting quantum atoms study of noncovalent interactions in the NCF⋯NH 3 complex
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2020-04-19 , DOI: 10.1002/jcc.26202 Nasim Orangi , Kiamars Eskandari 1
Journal of Computational Chemistry ( IF 3 ) Pub Date : 2020-04-19 , DOI: 10.1002/jcc.26202 Nasim Orangi , Kiamars Eskandari 1
Affiliation
|
Different computational methods are used to investigate the nature of interaction in the NCF⋯NH3 model complex, in which the fluorine atom acts as a Lewis acid and forms a noncovalent bond with the ammonia (Lewis base). Symmetry‐adapted perturbation theory based on density functional theory (SAPT(DFT)) indicates that the noncovalent interaction in the NCF⋯NH3 complex is mainly electrostatics. However, dispersion and induction terms also play important roles. Although fluorine noncovalent interactions are typically classified as halogen bonds, they are somewhat different from the well‐known halogen bonds of iodine, bromine, and chlorine. The halogen bonds of NCCl⋯NH3 and NCBr⋯NH3 are directional and the C X N (X = Cl or Br) angle tends to be linear. In contrast, the fluorine interaction in NCF⋯NH3 is not directional; the interaction energy shows no sensitivity to the angular (C F N) distortions, and the energy profile is flat over a wide angular range (from 180° to about 140°). However, for the angles less than 130°, the energy curve shows a clear angular dependence and the interaction between NCF and NH3 becomes stronger as the C F N angle decreases. It seems that at the tighter angles, a tetrel‐bonded NCF⋯NH3 complex is preferred. Moreover, interacting quantum atoms (IQA) analysis shows that the competition between different intra‐atomic and interatomic interactions determines the geometry of NCF⋯NH3 complex.
中文翻译:

氟作为路易斯酸:基于密度泛函理论和相互作用量子原子研究 NCF⋯NH 3 复合物中非共价相互作用的对称微扰理论
使用不同的计算方法来研究 NCF⋯NH3 模型复合物中相互作用的性质,其中氟原子充当路易斯酸并与氨(路易斯碱)形成非共价键。基于密度泛函理论 (SAPT(DFT)) 的对称适应扰动理论表明 NCF⋯NH3 复合物中的非共价相互作用主要是静电。然而,色散和归纳项也起着重要作用。尽管氟非共价相互作用通常被归类为卤素键,但它们与众所周知的碘、溴和氯的卤素键有些不同。NCCl⋯NH3 和NCBr⋯NH3 的卤键是定向的,C X N(X = Cl 或Br)角趋于线性。相比之下,NCF⋯NH3 中的氟相互作用不是定向的;相互作用能量对角度 (C F N) 失真不敏感,并且能量分布在很宽的角度范围内(从 180° 到约 140°)是平坦的。然而,对于小于 130°的角度,能量曲线显示出明显的角度依赖性,并且随着 C F N 角度的减小,NCF 和 NH3 之间的相互作用变得更强。似乎在更小的角度上,四角键合的 NCF⋯NH3 复合物是首选。此外,相互作用量子原子(IQA)分析表明,不同原子内和原子间相互作用之间的竞争决定了 NCF⋯NH3 复合物的几何形状。能量曲线显示出明显的角度依赖性,并且随着 C F N 角度的减小,NCF 和 NH3 之间的相互作用变得更强。似乎在更小的角度上,四角键合的 NCF⋯NH3 复合物是首选。此外,相互作用量子原子(IQA)分析表明,不同原子内和原子间相互作用之间的竞争决定了 NCF⋯NH3 复合物的几何形状。能量曲线显示出明显的角度依赖性,并且随着 C F N 角度的减小,NCF 和 NH3 之间的相互作用变得更强。似乎在更小的角度上,四角键合的 NCF⋯NH3 复合物是首选。此外,相互作用量子原子(IQA)分析表明,不同原子内和原子间相互作用之间的竞争决定了 NCF⋯NH3 复合物的几何形状。
更新日期:2020-04-19
中文翻译:
氟作为路易斯酸:基于密度泛函理论和相互作用量子原子研究 NCF⋯NH 3 复合物中非共价相互作用的对称微扰理论
使用不同的计算方法来研究 NCF⋯NH3 模型复合物中相互作用的性质,其中氟原子充当路易斯酸并与氨(路易斯碱)形成非共价键。基于密度泛函理论 (SAPT(DFT)) 的对称适应扰动理论表明 NCF⋯NH3 复合物中的非共价相互作用主要是静电。然而,色散和归纳项也起着重要作用。尽管氟非共价相互作用通常被归类为卤素键,但它们与众所周知的碘、溴和氯的卤素键有些不同。NCCl⋯NH3 和NCBr⋯NH3 的卤键是定向的,C X N(X = Cl 或Br)角趋于线性。相比之下,NCF⋯NH3 中的氟相互作用不是定向的;相互作用能量对角度 (C F N) 失真不敏感,并且能量分布在很宽的角度范围内(从 180° 到约 140°)是平坦的。然而,对于小于 130°的角度,能量曲线显示出明显的角度依赖性,并且随着 C F N 角度的减小,NCF 和 NH3 之间的相互作用变得更强。似乎在更小的角度上,四角键合的 NCF⋯NH3 复合物是首选。此外,相互作用量子原子(IQA)分析表明,不同原子内和原子间相互作用之间的竞争决定了 NCF⋯NH3 复合物的几何形状。能量曲线显示出明显的角度依赖性,并且随着 C F N 角度的减小,NCF 和 NH3 之间的相互作用变得更强。似乎在更小的角度上,四角键合的 NCF⋯NH3 复合物是首选。此外,相互作用量子原子(IQA)分析表明,不同原子内和原子间相互作用之间的竞争决定了 NCF⋯NH3 复合物的几何形状。能量曲线显示出明显的角度依赖性,并且随着 C F N 角度的减小,NCF 和 NH3 之间的相互作用变得更强。似乎在更小的角度上,四角键合的 NCF⋯NH3 复合物是首选。此外,相互作用量子原子(IQA)分析表明,不同原子内和原子间相互作用之间的竞争决定了 NCF⋯NH3 复合物的几何形状。


























 京公网安备 11010802027423号
京公网安备 11010802027423号