当前位置:
X-MOL 学术
›
Hum. Mutat.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
LEAP: Using machine learning to support variant classification in a clinical setting.
Human Mutation ( IF 3.9 ) Pub Date : 2020-04-01 , DOI: 10.1002/humu.24011 Carmen Lai 1 , Anjali D Zimmer 2 , Robert O'Connor 3 , Serra Kim 3 , Ray Chan 3 , Jeroen van den Akker 4 , Alicia Y Zhou 2 , Scott Topper 5 , Gilad Mishne 1
Human Mutation ( IF 3.9 ) Pub Date : 2020-04-01 , DOI: 10.1002/humu.24011 Carmen Lai 1 , Anjali D Zimmer 2 , Robert O'Connor 3 , Serra Kim 3 , Ray Chan 3 , Jeroen van den Akker 4 , Alicia Y Zhou 2 , Scott Topper 5 , Gilad Mishne 1
Affiliation
|
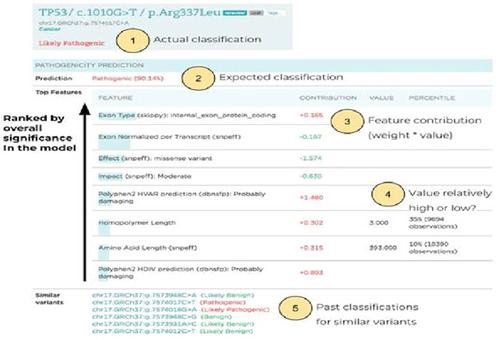
Advances in genome sequencing have led to a tremendous increase in the discovery of novel missense variants, but evidence for determining clinical significance can be limited or conflicting. Here, we present Learning from Evidence to Assess Pathogenicity (LEAP), a machine learning model that utilizes a variety of feature categories to classify variants, and achieves high performance in multiple genes and different health conditions. Feature categories include functional predictions, splice predictions, population frequencies, conservation scores, protein domain data, and clinical observation data such as personal and family history and covariant information. L2-regularized logistic regression and random forest classification models were trained on missense variants detected and classified during the course of routine clinical testing at Color Genomics (14,226 variants from 24 cancer-related genes and 5,398 variants from 30 cardiovascular-related genes). Using 10-fold cross-validated predictions, the logistic regression model achieved an area under the receiver operating characteristic curve (AUROC) of 97.8% (cancer) and 98.8% (cardiovascular), while the random forest model achieved 98.3% (cancer) and 98.6% (cardiovascular). We demonstrate generalizability to different genes by validating predictions on genes withheld from training (96.8% AUROC). High accuracy and broad applicability make LEAP effective in the clinical setting as a high-throughput quality control layer.
中文翻译:

LEAP:使用机器学习支持临床环境中的变异分类。
基因组测序的进步导致新错义变异的发现大幅增加,但确定临床意义的证据可能有限或相互矛盾。在这里,我们提出了从证据到评估致病性 (LEAP) 的学习,这是一种利用各种特征类别对变异进行分类的机器学习模型,并在多个基因和不同的健康状况下实现了高性能。特征类别包括功能预测、剪接预测、种群频率、保护分数、蛋白质域数据和临床观察数据,例如个人和家族史以及协变信息。L2 正则化逻辑回归和随机森林分类模型在 Color Genomics 常规临床测试过程中检测和分类的错义变异(来自 24 个癌症相关基因的 14,226 个变异和来自 30 个心血管相关基因的 5,398 个变异)进行了训练。使用 10 倍交叉验证预测,逻辑回归模型实现了 97.8%(癌症)和 98.8%(心血管)的受试者工作特征曲线(AUROC)下面积,而随机森林模型实现了 98.3%(癌症)和98.6%(心血管)。我们通过验证对训练中保留的基因的预测 (96.8% AUROC) 来证明对不同基因的普遍性。高精度和广泛的适用性使 LEAP 在临床环境中作为高通量质量控制层有效。24 个癌症相关基因的 226 个变异和 30 个心血管相关基因的 5,398 个变异)。使用 10 倍交叉验证预测,逻辑回归模型实现了 97.8%(癌症)和 98.8%(心血管)的受试者工作特征曲线(AUROC)下面积,而随机森林模型实现了 98.3%(癌症)和98.6%(心血管)。我们通过验证对训练中保留的基因的预测 (96.8% AUROC) 来证明对不同基因的普遍性。高精度和广泛的适用性使 LEAP 在临床环境中作为高通量质量控制层有效。24 个癌症相关基因的 226 个变异和 30 个心血管相关基因的 5,398 个变异)。使用 10 倍交叉验证预测,逻辑回归模型实现了 97.8%(癌症)和 98.8%(心血管)的受试者工作特征曲线(AUROC)下面积,而随机森林模型实现了 98.3%(癌症)和98.6%(心血管)。我们通过验证对训练中保留的基因的预测 (96.8% AUROC) 来证明对不同基因的普遍性。高精度和广泛的适用性使 LEAP 在临床环境中作为高通量质量控制层有效。8%(癌症)和 98.8%(心血管),而随机森林模型达到 98.3%(癌症)和 98.6%(心血管)。我们通过验证对训练中保留的基因的预测 (96.8% AUROC) 来证明对不同基因的普遍性。高精度和广泛的适用性使 LEAP 在临床环境中作为高通量质量控制层有效。8%(癌症)和 98.8%(心血管),而随机森林模型达到 98.3%(癌症)和 98.6%(心血管)。我们通过验证对训练中保留的基因的预测 (96.8% AUROC) 来证明对不同基因的普遍性。高精度和广泛的适用性使 LEAP 在临床环境中作为高通量质量控制层有效。
更新日期:2020-04-01
中文翻译:
LEAP:使用机器学习支持临床环境中的变异分类。
基因组测序的进步导致新错义变异的发现大幅增加,但确定临床意义的证据可能有限或相互矛盾。在这里,我们提出了从证据到评估致病性 (LEAP) 的学习,这是一种利用各种特征类别对变异进行分类的机器学习模型,并在多个基因和不同的健康状况下实现了高性能。特征类别包括功能预测、剪接预测、种群频率、保护分数、蛋白质域数据和临床观察数据,例如个人和家族史以及协变信息。L2 正则化逻辑回归和随机森林分类模型在 Color Genomics 常规临床测试过程中检测和分类的错义变异(来自 24 个癌症相关基因的 14,226 个变异和来自 30 个心血管相关基因的 5,398 个变异)进行了训练。使用 10 倍交叉验证预测,逻辑回归模型实现了 97.8%(癌症)和 98.8%(心血管)的受试者工作特征曲线(AUROC)下面积,而随机森林模型实现了 98.3%(癌症)和98.6%(心血管)。我们通过验证对训练中保留的基因的预测 (96.8% AUROC) 来证明对不同基因的普遍性。高精度和广泛的适用性使 LEAP 在临床环境中作为高通量质量控制层有效。24 个癌症相关基因的 226 个变异和 30 个心血管相关基因的 5,398 个变异)。使用 10 倍交叉验证预测,逻辑回归模型实现了 97.8%(癌症)和 98.8%(心血管)的受试者工作特征曲线(AUROC)下面积,而随机森林模型实现了 98.3%(癌症)和98.6%(心血管)。我们通过验证对训练中保留的基因的预测 (96.8% AUROC) 来证明对不同基因的普遍性。高精度和广泛的适用性使 LEAP 在临床环境中作为高通量质量控制层有效。24 个癌症相关基因的 226 个变异和 30 个心血管相关基因的 5,398 个变异)。使用 10 倍交叉验证预测,逻辑回归模型实现了 97.8%(癌症)和 98.8%(心血管)的受试者工作特征曲线(AUROC)下面积,而随机森林模型实现了 98.3%(癌症)和98.6%(心血管)。我们通过验证对训练中保留的基因的预测 (96.8% AUROC) 来证明对不同基因的普遍性。高精度和广泛的适用性使 LEAP 在临床环境中作为高通量质量控制层有效。8%(癌症)和 98.8%(心血管),而随机森林模型达到 98.3%(癌症)和 98.6%(心血管)。我们通过验证对训练中保留的基因的预测 (96.8% AUROC) 来证明对不同基因的普遍性。高精度和广泛的适用性使 LEAP 在临床环境中作为高通量质量控制层有效。8%(癌症)和 98.8%(心血管),而随机森林模型达到 98.3%(癌症)和 98.6%(心血管)。我们通过验证对训练中保留的基因的预测 (96.8% AUROC) 来证明对不同基因的普遍性。高精度和广泛的适用性使 LEAP 在临床环境中作为高通量质量控制层有效。


























 京公网安备 11010802027423号
京公网安备 11010802027423号