Nature Biotechnology ( IF 46.9 ) Pub Date : 2020-04-06 , DOI: 10.1038/s41587-020-0469-4 Elisabetta Mereu 1 , Atefeh Lafzi 1 , Catia Moutinho 1 , Christoph Ziegenhain 2 , Davis J McCarthy 3, 4, 5 , Adrián Álvarez-Varela 6 , Eduard Batlle 6, 7, 8 , Sagar 9 , Dominic Grün 9 , Julia K Lau 10 , Stéphane C Boutet 10 , Chad Sanada 11 , Aik Ooi 11 , Robert C Jones 12 , Kelly Kaihara 13 , Chris Brampton 13 , Yasha Talaga 13 , Yohei Sasagawa 14 , Kaori Tanaka 14 , Tetsutaro Hayashi 14 , Caroline Braeuning 15 , Cornelius Fischer 15 , Sascha Sauer 15 , Timo Trefzer 16 , Christian Conrad 16 , Xian Adiconis 17, 18 , Lan T Nguyen 17 , Aviv Regev 17, 19, 20 , Joshua Z Levin 17, 18 , Swati Parekh 21 , Aleksandar Janjic 22 , Lucas E Wange 22 , Johannes W Bagnoli 22 , Wolfgang Enard 22 , Marta Gut 1 , Rickard Sandberg 2 , Itoshi Nikaido 14, 23 , Ivo Gut 1, 24 , Oliver Stegle 3, 4, 25 , Holger Heyn 1, 24
|
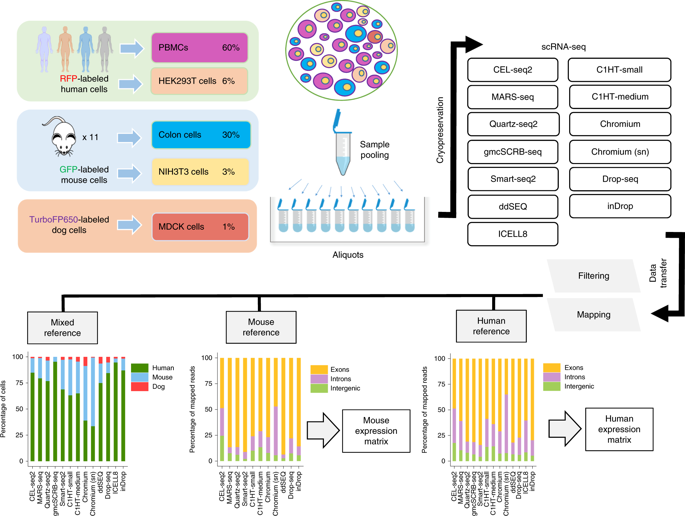
Single-cell RNA sequencing (scRNA-seq) is the leading technique for characterizing the transcriptomes of individual cells in a sample. The latest protocols are scalable to thousands of cells and are being used to compile cell atlases of tissues, organs and organisms. However, the protocols differ substantially with respect to their RNA capture efficiency, bias, scale and costs, and their relative advantages for different applications are unclear. In the present study, we generated benchmark datasets to systematically evaluate protocols in terms of their power to comprehensively describe cell types and states. We performed a multicenter study comparing 13 commonly used scRNA-seq and single-nucleus RNA-seq protocols applied to a heterogeneous reference sample resource. Comparative analysis revealed marked differences in protocol performance. The protocols differed in library complexity and their ability to detect cell-type markers, impacting their predictive value and suitability for integration into reference cell atlases. These results provide guidance both for individual researchers and for consortium projects such as the Human Cell Atlas.
中文翻译:
对用于细胞图集项目的单细胞RNA测序协议进行基准测试。
单细胞RNA测序(scRNA-seq)是表征样品中单个细胞的转录组的主要技术。最新的协议可扩展到数千个细胞,并被用于编译组织,器官和生物体的细胞图集。但是,协议在RNA捕获效率,偏倚,规模和成本方面有很大不同,并且它们在不同应用中的相对优势尚不清楚。在本研究中,我们生成了基准数据集以根据其全面描述细胞类型和状态的能力来系统评估协议。我们进行了一项多中心研究,比较了13种常用scRNA-seq和单核RNA-seq方案应用于异质参考样品资源。比较分析显示协议性能存在明显差异。这些协议在库的复杂性和检测细胞类型标记的能力方面有所不同,从而影响了它们的预测价值和与参考细胞图谱整合的适用性。这些结果为个人研究人员和诸如人类细胞图谱等财团项目提供了指导。
























 京公网安备 11010802027423号
京公网安备 11010802027423号