当前位置:
X-MOL 学术
›
J. Hepatol.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
MLKL-dependent signaling regulates autophagic flux in a murine model of non-alcoholic fatty liver disease
Journal of Hepatology ( IF 25.7 ) Pub Date : 2020-09-01 , DOI: 10.1016/j.jhep.2020.03.023 Xiaoqin Wu 1 , Kyle L Poulsen 1 , Carlos Sanz-Garcia 1 , Emily Huang 1 , Megan R McMullen 1 , Sanjoy Roychowdhury 2 , Srinivasan Dasarathy 3 , Laura E Nagy 3
Journal of Hepatology ( IF 25.7 ) Pub Date : 2020-09-01 , DOI: 10.1016/j.jhep.2020.03.023 Xiaoqin Wu 1 , Kyle L Poulsen 1 , Carlos Sanz-Garcia 1 , Emily Huang 1 , Megan R McMullen 1 , Sanjoy Roychowdhury 2 , Srinivasan Dasarathy 3 , Laura E Nagy 3
Affiliation
|
BACKGROUND & AIMS
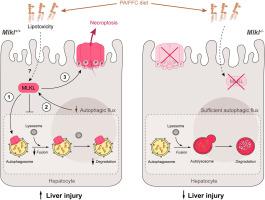
Autophagy maintains cellular homeostasis and plays a critical role in the development of non-alcohol-related liver disease (NAFL/NASH). Insufficient autophagy can lead to hepatocellular injury and death. The pseudokinase mixed lineage kinase domain-like (MLKL) is a key downstream effector of receptor interacting protein kinase 3 (RIP3) in the necroptotic pathway of programmed cell death. However, recent data reveal that MLKL also regulates autophagy. Here we tested the hypothesis that MLKL contributes to the progression of Western diet-induced liver injury in mice by regulating autophagy. METHODS
Rip3+/+, Rip3-/-, Mlkl+/+ and Mlkl-/- mice were fed Western diet (FFC diet, high in fat, fructose and cholesterol) or chow for 12 weeks. AML12 and primary mouse hepatocytes were exposed to palmitic acid (PA). RESULTS
FFC diet increased expression, phosphorylation and oligomerization of MLKL in the liver. Mlkl, but not Rip3, deficiency, protected mice from FFC diet-induced liver injury. FFC diet also induced accumulation of p62 and LC3-II, as well as markers of ER stress, in Mlkl+/+, but not Mlkl-/- mice. Mlkl deficiency in mice also prevented the inhibition of autophagy by a protease inhibitor, leupeptin. Using a mRFP-GFP-LC3 reporter in cultured hepatocytes revealed that PA blocked the fusion of autophagosomes with lysosomes. PA triggered MLKL expression and translocation to autophagosomes prior to plasma membrane independently of Rip3. Mlkl, but not Rip3, deficiency prevented inhibition of autophagy in PA-treated hepatocytes. Overexpression of Mlkl blocked autophagy independently of PA. Additionally, pharmacologic inhibition of autophagy induced MLKL expression and translocation to plasma membrane in hepatocytes. CONCLUSIONS
Taken together, these data indicate that MLKL-dependent, but RIP3-independent, signaling contributes to FFC diet-induced liver injury through inhibition of autophagy.
中文翻译:

MLKL 依赖性信号调节非酒精性脂肪性肝病小鼠模型中的自噬通量
背景和目的 自噬维持细胞稳态,并在非酒精相关性肝病 (NAFL/NASH) 的发展中发挥关键作用。自噬不足会导致肝细胞损伤和死亡。假激酶混合谱系激酶域样 (MLKL) 是受体相互作用蛋白激酶 3 (RIP3) 在程序性细胞死亡的坏死性途径中的关键下游效应器。然而,最近的数据显示 MLKL 也调节自噬。在这里,我们测试了 MLKL 通过调节自噬促进西方饮食诱导的小鼠肝损伤进展的假设。方法 Rip3+/+、Rip3-/-、Mlkl+/+ 和 Mlkl-/- 小鼠喂食西方饮食(FFC 饮食,高脂肪、果糖和胆固醇)或食物 12 周。AML12 和原代小鼠肝细胞暴露于棕榈酸 (PA)。结果 FFC 饮食增加了肝脏中 MLKL 的表达、磷酸化和寡聚化。Mlk1,但不是 Rip3,缺乏保护小鼠免受 FFC 饮食诱导的肝损伤。FFC 饮食还在 Mlkl+/+ 而非 Mlkl-/- 小鼠中诱导 p62 和 LC3-II 以及 ER 应激标志物的积累。小鼠中的 Mlkl 缺乏也阻止了蛋白酶抑制剂亮肽素对自噬的抑制。在培养的肝细胞中使用 mRFP-GFP-LC3 报告基因显示 PA 阻断了自噬体与溶酶体的融合。PA 独立于 Rip3 在质膜之前触发 MLKL 表达和易位至自噬体。Mlk1,但不是 Rip3,缺陷阻止了 PA 处理的肝细胞中自噬的抑制。Mlk1的过表达独立于PA阻断自噬。此外,药理抑制自噬诱导的 MLKL 表达和肝细胞质膜易位。结论 综上所述,这些数据表明依赖于 MLKL 但不依赖于 RIP3 的信号通过抑制自噬导致 FFC 饮食诱导的肝损伤。
更新日期:2020-09-01
中文翻译:
MLKL 依赖性信号调节非酒精性脂肪性肝病小鼠模型中的自噬通量
背景和目的 自噬维持细胞稳态,并在非酒精相关性肝病 (NAFL/NASH) 的发展中发挥关键作用。自噬不足会导致肝细胞损伤和死亡。假激酶混合谱系激酶域样 (MLKL) 是受体相互作用蛋白激酶 3 (RIP3) 在程序性细胞死亡的坏死性途径中的关键下游效应器。然而,最近的数据显示 MLKL 也调节自噬。在这里,我们测试了 MLKL 通过调节自噬促进西方饮食诱导的小鼠肝损伤进展的假设。方法 Rip3+/+、Rip3-/-、Mlkl+/+ 和 Mlkl-/- 小鼠喂食西方饮食(FFC 饮食,高脂肪、果糖和胆固醇)或食物 12 周。AML12 和原代小鼠肝细胞暴露于棕榈酸 (PA)。结果 FFC 饮食增加了肝脏中 MLKL 的表达、磷酸化和寡聚化。Mlk1,但不是 Rip3,缺乏保护小鼠免受 FFC 饮食诱导的肝损伤。FFC 饮食还在 Mlkl+/+ 而非 Mlkl-/- 小鼠中诱导 p62 和 LC3-II 以及 ER 应激标志物的积累。小鼠中的 Mlkl 缺乏也阻止了蛋白酶抑制剂亮肽素对自噬的抑制。在培养的肝细胞中使用 mRFP-GFP-LC3 报告基因显示 PA 阻断了自噬体与溶酶体的融合。PA 独立于 Rip3 在质膜之前触发 MLKL 表达和易位至自噬体。Mlk1,但不是 Rip3,缺陷阻止了 PA 处理的肝细胞中自噬的抑制。Mlk1的过表达独立于PA阻断自噬。此外,药理抑制自噬诱导的 MLKL 表达和肝细胞质膜易位。结论 综上所述,这些数据表明依赖于 MLKL 但不依赖于 RIP3 的信号通过抑制自噬导致 FFC 饮食诱导的肝损伤。


























 京公网安备 11010802027423号
京公网安备 11010802027423号