当前位置:
X-MOL 学术
›
J. Proteomics
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
An experimentally generated peptide database increases the sensitivity of XL-MS with complex samples.
Journal of Proteomics ( IF 3.3 ) Pub Date : 2020-03-19 , DOI: 10.1016/j.jprot.2020.103754 Iwan Parfentev 1 , Sandra Schilbach 2 , Patrick Cramer 2 , Henning Urlaub 3
Journal of Proteomics ( IF 3.3 ) Pub Date : 2020-03-19 , DOI: 10.1016/j.jprot.2020.103754 Iwan Parfentev 1 , Sandra Schilbach 2 , Patrick Cramer 2 , Henning Urlaub 3
Affiliation
|
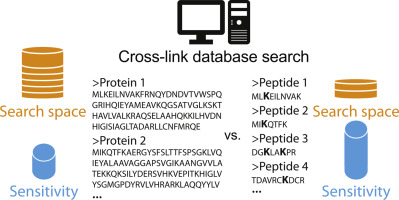
Cross-linking mass spectrometry (XL-MS) is steadily expanding its range of applications from purified protein complexes to more complex samples like organelles and even entire cells. One main challenge using non-cleavable cross-linkers is the so-called n2 problem: With linearly increasing database size, the search space for the identification of two covalently linked peptides per spectrum increases quadratically. Here, we report an alternative search strategy that focuses on only those peptides, which were demonstrated to cross-link under the applied experimental conditions. The performance of a parallel XL-MS experiment using a thiol-cleavable cross-linker enabled the identification of peptides that carried a cleaved cross-link moiety after reduction and hence were involved in cross-linking reactions. Based on these identifications, a peptide database was generated and used for the database search of the actual cross-linking experiment with a non-cleavable cross-linker. This peptide-focused approach was tested on protein complexes with a reported structural model and obtained results corresponded well to a conventional database search. An application of the strategy on in vivo cross-linked Bacillus subtilis and Bacillus cereus cells revealed a five- to tenfold reduction in search time and led to significantly more identifications with the latter species than a search against the entire proteome. SIGNIFICANCE: Instead of considering all theoretically cross-linkable peptides in a proteome, identification and pre-filtering for a subset of cross-link peptide candidates allows for a dramatically decreased search space. Hence, there is less potential for the random accumulation of false positives ultimately leading to a higher sensitivity in the XL-MS experiment. Using the peptide-focused approach, a cross-linking database search can be conducted in a fraction of time while yielding a similar or higher number of identifications, thereby enabling the cross-linking analysis of samples of mammalian proteome complexity.
中文翻译:

实验生成的肽数据库提高了 XL-MS 对复杂样品的灵敏度。
交联质谱 (XL-MS) 正在稳步扩大其应用范围,从纯化的蛋白质复合物到更复杂的样品,如细胞器甚至整个细胞。使用不可切割交联剂的一个主要挑战是所谓的 n2 问题:随着数据库大小的线性增加,用于识别每个光谱的两个共价连接肽的搜索空间呈二次增加。在这里,我们报告了一种替代搜索策略,该策略仅关注那些在应用的实验条件下被证明可以交联的肽。使用硫醇可裂解交联剂的平行 XL-MS 实验的性能能够鉴定在还原后带有裂解交联部分并因此参与交联反应的肽。根据这些鉴定,生成了一个肽数据库,并将其用于具有不可切割交联剂的实际交联实验的数据库搜索。这种以肽为中心的方法在具有报告结构模型的蛋白质复合物上进行了测试,获得的结果与传统的数据库搜索非常吻合。该策略在体内交联枯草芽孢杆菌和蜡状芽孢杆菌细胞上的应用显示,搜索时间减少了 5 到 10 倍,并且与对整个蛋白质组的搜索相比,对后者的识别明显更多。意义:不是考虑蛋白质组中所有理论上可交联的肽,而是对一部分交联肽候选物进行识别和预过滤,从而显着减少搜索空间。因此,随机累积假阳性的可能性较小,最终导致 XL-MS 实验的灵敏度更高。使用以肽为中心的方法,交联数据库搜索可以在很短的时间内进行,同时产生相似或更多数量的鉴定,从而能够对哺乳动物蛋白质组复杂的样本进行交联分析。
更新日期:2020-03-20
中文翻译:
实验生成的肽数据库提高了 XL-MS 对复杂样品的灵敏度。
交联质谱 (XL-MS) 正在稳步扩大其应用范围,从纯化的蛋白质复合物到更复杂的样品,如细胞器甚至整个细胞。使用不可切割交联剂的一个主要挑战是所谓的 n2 问题:随着数据库大小的线性增加,用于识别每个光谱的两个共价连接肽的搜索空间呈二次增加。在这里,我们报告了一种替代搜索策略,该策略仅关注那些在应用的实验条件下被证明可以交联的肽。使用硫醇可裂解交联剂的平行 XL-MS 实验的性能能够鉴定在还原后带有裂解交联部分并因此参与交联反应的肽。根据这些鉴定,生成了一个肽数据库,并将其用于具有不可切割交联剂的实际交联实验的数据库搜索。这种以肽为中心的方法在具有报告结构模型的蛋白质复合物上进行了测试,获得的结果与传统的数据库搜索非常吻合。该策略在体内交联枯草芽孢杆菌和蜡状芽孢杆菌细胞上的应用显示,搜索时间减少了 5 到 10 倍,并且与对整个蛋白质组的搜索相比,对后者的识别明显更多。意义:不是考虑蛋白质组中所有理论上可交联的肽,而是对一部分交联肽候选物进行识别和预过滤,从而显着减少搜索空间。因此,随机累积假阳性的可能性较小,最终导致 XL-MS 实验的灵敏度更高。使用以肽为中心的方法,交联数据库搜索可以在很短的时间内进行,同时产生相似或更多数量的鉴定,从而能够对哺乳动物蛋白质组复杂的样本进行交联分析。


























 京公网安备 11010802027423号
京公网安备 11010802027423号