当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Hydrogen Adsorption and Dissociation on AlnRh2+ (n = 1 to 9) Clusters: Steric and Coordination Effects
The Journal of Physical Chemistry C ( IF 3.7 ) Pub Date : 2020-03-27 , DOI: 10.1021/acs.jpcc.9b11230 Meiye Jia 1 , Jan Vanbuel 1 , Piero Ferrari 1 , Wieland Schöllkopf 2 , André Fielicke 2 , Minh Tho Nguyen 3 , Ewald Janssens 1
The Journal of Physical Chemistry C ( IF 3.7 ) Pub Date : 2020-03-27 , DOI: 10.1021/acs.jpcc.9b11230 Meiye Jia 1 , Jan Vanbuel 1 , Piero Ferrari 1 , Wieland Schöllkopf 2 , André Fielicke 2 , Minh Tho Nguyen 3 , Ewald Janssens 1
Affiliation
|
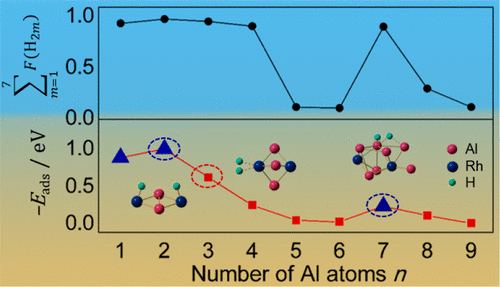
The interaction of molecular hydrogen with doubly rhodium doped aluminum clusters, AlnRh2+ (n = 1 to 9), is investigated by a combination of time-of-flight mass spectrometry, infrared multiple photon dissociation spectroscopy, and density functional theory calculations. The reactivity of the AlnRh2+ clusters toward H2 is found to be sensitive to cluster size, with sizes n = 1 to 4 and 7 being the most reactive. Al3Rh2+ and Al4Rh2+ are the only species that thermodynamically prefer molecular over dissociative H2 adsorption. Calculated molecular adsorption energies of a single H2 molecule correlate well with the experimental abundances of the hydrogenated species, and the potential energy profiles reveal that H2 dissociation only has submerged barriers for n = 1, 2, and 7. In contrast, the molecularly hydrogenated complexes seem to be kinetically trapped for n = 5, 6, 8, and 9 due to significant energy barriers. This indicates that the initial molecular H2 adsorption on the Rh atoms and thereafter dissociation are the determining steps for the hydrogenation reaction. An analysis of the cluster geometries reveals that the coordination environment and the steric factor of the Rh atoms are the main descriptors for the size-dependent reactivity of the AlnRh2+ clusters.
中文翻译:

Al n Rh 2 +(n = 1至9)团簇上的氢吸附和解离:立体效应和配位效应
通过飞行时间质谱,红外多光子解离光谱和密度泛函理论计算的组合,研究了分子氢与双铑掺杂的铝团簇Al n Rh 2 +(n = 1至9)的相互作用。。发现Al n Rh 2 +团簇对H 2的反应性对团簇尺寸敏感,尺寸n = 1-4至4和7是最具反应性的。Al 3 Rh 2 +和Al 4 Rh 2 +是热力学上唯一喜欢分子而不是离解H 2吸附的物种。计算得出的单个H 2分子的分子吸附能与氢化物种的实验丰度有很好的相关性,并且势能图显示H 2的解离仅具有n = 1、2和7的浸没势垒。由于明显的能垒,氢化复合物似乎在动力学上被困为n = 5、6、8和9。这表明初始分子H 2吸附在Rh原子上,然后解离是氢化反应的决定性步骤。对团簇几何形状的分析表明,Rh原子的配位环境和空间因子是Al n Rh 2 +团簇的尺寸依赖性反应性的主要描述子。
更新日期:2020-03-27
中文翻译:
Al n Rh 2 +(n = 1至9)团簇上的氢吸附和解离:立体效应和配位效应
通过飞行时间质谱,红外多光子解离光谱和密度泛函理论计算的组合,研究了分子氢与双铑掺杂的铝团簇Al n Rh 2 +(n = 1至9)的相互作用。。发现Al n Rh 2 +团簇对H 2的反应性对团簇尺寸敏感,尺寸n = 1-4至4和7是最具反应性的。Al 3 Rh 2 +和Al 4 Rh 2 +是热力学上唯一喜欢分子而不是离解H 2吸附的物种。计算得出的单个H 2分子的分子吸附能与氢化物种的实验丰度有很好的相关性,并且势能图显示H 2的解离仅具有n = 1、2和7的浸没势垒。由于明显的能垒,氢化复合物似乎在动力学上被困为n = 5、6、8和9。这表明初始分子H 2吸附在Rh原子上,然后解离是氢化反应的决定性步骤。对团簇几何形状的分析表明,Rh原子的配位环境和空间因子是Al n Rh 2 +团簇的尺寸依赖性反应性的主要描述子。

























 京公网安备 11010802027423号
京公网安备 11010802027423号