当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Fragment Pose Prediction Using Non-equilibrium Candidate Monte Carlo and Molecular Dynamics Simulations.
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2020-03-13 , DOI: 10.1021/acs.jctc.9b01096 Nathan M Lim 1 , Meghan Osato 1 , Gregory L Warren 2 , David L Mobley 1, 3
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2020-03-13 , DOI: 10.1021/acs.jctc.9b01096 Nathan M Lim 1 , Meghan Osato 1 , Gregory L Warren 2 , David L Mobley 1, 3
Affiliation
|
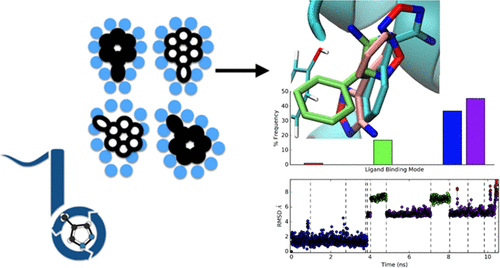
Part of early stage drug discovery involves determining how molecules may bind to the target protein. Through understanding where and how molecules bind, chemists can begin to build ideas on how to design improvements to increase binding affinities. In this retrospective study, we compare how computational approaches like docking, molecular dynamics (MD) simulations, and a non-equilibrium candidate Monte Carlo (NCMC)-based method (NCMC + MD) perform in predicting binding modes for a set of 12 fragment-like molecules, which bind to soluble epoxide hydrolase. We evaluate each method’s effectiveness in identifying the dominant binding mode and finding additional binding modes (if any). Then, we compare our predicted binding modes to experimentally obtained X-ray crystal structures. We dock each of the 12 small molecules into the apo-protein crystal structure and then run simulations up to 1 μs each. Small and fragment-like molecules likely have smaller energy barriers separating different binding modes by virtue of relatively fewer and weaker interactions relative to drug-like molecules and thus likely undergo more rapid binding mode transitions. We expect, thus, to see more rapid transitions between binding modes in our study. Following this, we build Markov State Models to define our stable ligand binding modes. We investigate if adequate sampling of ligand binding modes and transitions between them can occur at the microsecond timescale using traditional MD or a hybrid NCMC+MD simulation approach. Our findings suggest that even with small fragment-like molecules, we fail to sample all the crystallographic binding modes using microsecond MD simulations, but using NCMC+MD, we have better success in sampling the crystal structure while obtaining the correct populations.
中文翻译:

使用非平衡候选蒙特卡洛和分子动力学模拟的片段姿势预测。
早期药物发现的一部分涉及确定分子如何与靶蛋白结合。通过了解分子结合的位置和方式,化学家可以开始构想如何设计改进方法以增加结合亲和力。在这项回顾性研究中,我们比较了对接,分子动力学(MD)模拟和非平衡候选基于蒙特卡洛(NCMC)的方法(NCMC + MD)等计算方法在预测一组12个片段的结合模式时如何执行类分子,与可溶性环氧化物水解酶结合。我们评估每种方法在识别主要结合模式和寻找其他结合模式(如果有)方面的有效性。然后,我们将预测的结合模式与实验获得的X射线晶体结构进行比较。我们将12个小分子中的每一个停靠在脱辅基蛋白晶体结构中,然后分别进行高达1μs的仿真。相对于药物样分子而言,相对较小且较弱的相互作用,小分子和碎片样分子可能具有较小的能垒,从而分隔不同的结合模式,因此可能经历更快的结合模式转变。因此,我们期望在我们的研究中看到绑定模式之间更快速的过渡。接下来,我们建立马尔可夫状态模型来定义我们稳定的配体结合模式。我们调查是否可以使用传统的MD或NCMC + MD混合仿真方法在微秒级的时间范围内对配体结合模式及其之间的过渡进行足够的采样。我们的发现表明,即使是碎片状小分子,
更新日期:2020-04-24
中文翻译:
使用非平衡候选蒙特卡洛和分子动力学模拟的片段姿势预测。
早期药物发现的一部分涉及确定分子如何与靶蛋白结合。通过了解分子结合的位置和方式,化学家可以开始构想如何设计改进方法以增加结合亲和力。在这项回顾性研究中,我们比较了对接,分子动力学(MD)模拟和非平衡候选基于蒙特卡洛(NCMC)的方法(NCMC + MD)等计算方法在预测一组12个片段的结合模式时如何执行类分子,与可溶性环氧化物水解酶结合。我们评估每种方法在识别主要结合模式和寻找其他结合模式(如果有)方面的有效性。然后,我们将预测的结合模式与实验获得的X射线晶体结构进行比较。我们将12个小分子中的每一个停靠在脱辅基蛋白晶体结构中,然后分别进行高达1μs的仿真。相对于药物样分子而言,相对较小且较弱的相互作用,小分子和碎片样分子可能具有较小的能垒,从而分隔不同的结合模式,因此可能经历更快的结合模式转变。因此,我们期望在我们的研究中看到绑定模式之间更快速的过渡。接下来,我们建立马尔可夫状态模型来定义我们稳定的配体结合模式。我们调查是否可以使用传统的MD或NCMC + MD混合仿真方法在微秒级的时间范围内对配体结合模式及其之间的过渡进行足够的采样。我们的发现表明,即使是碎片状小分子,
























 京公网安备 11010802027423号
京公网安备 11010802027423号