当前位置:
X-MOL 学术
›
Bioorgan. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
New 1,2,4-triazole/pyrazole hybrids linked to oxime moiety as nitric oxide donor celecoxib analogs: Synthesis, cyclooxygenase inhibition anti-inflammatory, ulcerogenicity, anti-proliferative activities, apoptosis, molecular modeling and nitric oxide release studies.
Bioorganic Chemistry ( IF 5.1 ) Pub Date : 2020-03-12 , DOI: 10.1016/j.bioorg.2020.103752 Wael A A Fadaly 1 , Yaseen A M M Elshaier 2 , Emad H M Hassanein 3 , Khaled R A Abdellatif 4
Bioorganic Chemistry ( IF 5.1 ) Pub Date : 2020-03-12 , DOI: 10.1016/j.bioorg.2020.103752 Wael A A Fadaly 1 , Yaseen A M M Elshaier 2 , Emad H M Hassanein 3 , Khaled R A Abdellatif 4
Affiliation
|
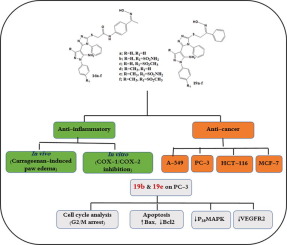
Two new series of hybrid structures 16a-f and 19a-f containing 1,2,4-triazole moiety, pyrazole core with COX-2 pharmacophore and oxime as NO donor moiety were designed, synthesized and evaluated for anti-inflammatory, cytotoxic activities and NO release. All compounds were more selective for COX-2 isozyme especially the sulphamoyl derivatives (16b, 16e, 19b and 19e) had COX-2 selectivity indexes (S.I. = 9.78, 8.57, 10.78 and 10.47 respectively) in comparison to celecoxib (S.I. = 8.68). Similarly, 16b, 16e, 19b and 19e were the most potent anti-inflammatory derivatives with ED50 = 46.98-54.45 μmol/kg better than celecoxib (ED50 = 76.09 μmol/kg). Also, 16b, 16e, 19b and 19e were significantly less ulcerogenic (ulcer indexes = 2.79-3.95) upon comparison with ibuprofen (ulcer index = 20.25) and comparable with celecoxib (ulcer index = 2.93). Regarding anti-cancer activity, most of the target derivatives 16a-f and 19a-f showed good activities against A-549, MCF-7, HCT-116 and PC-3 cancer cell lines. Additionally, these derivatives examined against F180 fibroblasts to investigate their selectivity indexes. The sulphamoyl derivatives with internal oxime 19b and 19e were the most potent derivatives against all used cell lines especially PC-3 (IC50 = 1.48 and 0.33 µM respectively) with 11.75 and 39.4-fold respectively selectivity towards PC-3 than F180 fibroblasts. The mechanistic investigation of 19b and 19e revealed that both compounds arrested cell cycle at G2/M phase by 32.16 and 39.95 folds, up-regulated Bax expression by 6.83 and 14.52 folds and down-regulated the expression of the gene Bcl-2 by 0.57 and 0.36fold respectively. Also, 19b and 19e were good inhibitor for p38MAPK (0.65 for 19b and 0.58 for 19e) and VEGFR-2 (0.39 for 19b and 0.54 for 19e) in comparison with PC-3 control cell. All compounds 16a-f and 19a-f released NO in a slow rate (0.15-3.17%) and the four sulphamoyl derivatives 16b, 16e, 19b and 19e were the most NO releasers (3.06, 2.15, 3.17 and 2.54% respectively). Docking studies were carried out to explain the interaction of 16a-f and 19a-f with the target enzymes. Docking mode of final designed compounds with celecoxib (ID: 3LN1) represented that their triazole ring adopted as the core aryl in Y shaped structure. Regarding EGFR inhibition, docking was carried out with ID: 1M17. The internal oxime serious was more active as anticancer because of their ability to form extra HBs with receptor cleft.
中文翻译:

与肟部分连接的新的1,2,4-三唑/吡唑杂物作为一氧化氮供体celecoxib类似物:合成,环氧合酶抑制抗炎,致溃疡性,抗增殖活性,细胞凋亡,分子模型和一氧化氮释放研究。
设计,合成并评估了两个新系列的杂合结构16a-f和19a-f,它们分别包含1,2,4-三唑部分,吡唑核心以及COX-2药效团和肟作为NO供体部分,并评估了其的抗炎,细胞毒性活性和没有释放。与塞来昔布(SI = 8.68)相比,所有化合物对COX-2同工酶的选择性更高,尤其是氨磺酰基衍生物(16b,16e,19b和19e)的COX-2选择性指数分别为(SI = 9.78、8.57、10.78和10.47)。 。同样,16b,16e,19b和19e是最有效的抗炎衍生物,ED50 =塞来昔布(ED50 = 76.09μmol/ kg),ED50 = 46.98-54.45μmol/ kg。同样,与布洛芬(溃疡指数= 20.25)相比,16b,16e,19b和19e的致溃疡性(溃疡指数= 2.79-3.95)明显更少,与塞来昔布(溃疡指数= 2.93)相当。关于抗癌活性,大多数目标衍生物16a-f和19a-f显示出对A-549,MCF-7,HCT-116和PC-3癌细胞系的良好活性。此外,这些衍生物针对F180成纤维细胞进行了研究,以研究其选择性指数。具有内部肟19b和19e的氨磺酰基衍生物是对所有使用过的细胞系最有效的衍生物,尤其是PC-3(分别为IC50 = 1.48和0.33 µM),对PC-3的选择性分别是F180成纤维细胞的11.75和39.4倍。对19b和19e的机理研究表明,这两种化合物在G2 / M期将细胞周期停滞了32.16和39.95倍,将Bax表达上调了6.83和14.52倍,并将基因Bcl-2的表达下调了0.57和。分别是0.36倍 同样,19b和19e是p38MAPK的良好抑制剂(0。与PC-3对照细胞相比,19b为65,19e为0.58)和VEGFR-2(19b为0.39,19e为0.54)。所有化合物16a-f和19a-f均以缓慢的速率(0.15-3.17%)释放NO,并且四种氨磺酰基衍生物16b,16e,19b和19e是最大的NO释放剂(分别为3.06、2.15、3.17和2.54%)。进行了对接研究以解释16a-f和19a-f与靶酶的相互作用。最终设计的化合物与塞来昔布(ID:3LN1)的对接模式表明,它们的三唑环被用作Y形结构中的核心芳基。关于EGFR抑制,以ID:1M17进行对接。严重的内部肟作为抗癌活性更高,因为它们能够与受体裂口形成额外的HBs。所有化合物16a-f和19a-f均以缓慢的速率(0.15-3.17%)释放NO,并且四种氨磺酰基衍生物16b,16e,19b和19e是最大的NO释放剂(分别为3.06、2.15、3.17和2.54%)。进行了对接研究以解释16a-f和19a-f与目标酶的相互作用。最终设计的化合物与塞来昔布(ID:3LN1)的对接模式表明,它们的三唑环被用作Y形结构中的核心芳基。关于EGFR抑制,以ID:1M17进行对接。严重的内部肟作为抗癌活性更高,因为它们能够与受体裂口形成额外的HBs。所有化合物16a-f和19a-f均以缓慢的速率(0.15-3.17%)释放NO,并且四种氨磺酰基衍生物16b,16e,19b和19e是最大的NO释放剂(分别为3.06、2.15、3.17和2.54%)。进行了对接研究以解释16a-f和19a-f与靶酶的相互作用。最终设计的化合物与塞来昔布(ID:3LN1)的对接模式表明,它们的三唑环被用作Y形结构中的核心芳基。关于EGFR抑制,以ID:1M17进行对接。严重的内部肟作为抗癌活性更高,因为它们能够与受体裂口形成额外的HBs。进行了对接研究以解释16a-f和19a-f与靶酶的相互作用。最终设计的化合物与塞来昔布(ID:3LN1)的对接模式表明,它们的三唑环被用作Y形结构中的核心芳基。关于EGFR抑制,以ID:1M17进行对接。严重的内部肟作为抗癌活性更高,因为它们能够与受体裂口形成额外的HBs。进行了对接研究以解释16a-f和19a-f与目标酶的相互作用。最终设计的化合物与塞来昔布(ID:3LN1)的对接模式表明,它们的三唑环被用作Y形结构中的核心芳基。关于EGFR抑制,以ID:1M17进行对接。严重的内部肟作为抗癌活性更高,因为它们能够与受体裂口形成额外的HBs。
更新日期:2020-03-12
中文翻译:
与肟部分连接的新的1,2,4-三唑/吡唑杂物作为一氧化氮供体celecoxib类似物:合成,环氧合酶抑制抗炎,致溃疡性,抗增殖活性,细胞凋亡,分子模型和一氧化氮释放研究。
设计,合成并评估了两个新系列的杂合结构16a-f和19a-f,它们分别包含1,2,4-三唑部分,吡唑核心以及COX-2药效团和肟作为NO供体部分,并评估了其的抗炎,细胞毒性活性和没有释放。与塞来昔布(SI = 8.68)相比,所有化合物对COX-2同工酶的选择性更高,尤其是氨磺酰基衍生物(16b,16e,19b和19e)的COX-2选择性指数分别为(SI = 9.78、8.57、10.78和10.47)。 。同样,16b,16e,19b和19e是最有效的抗炎衍生物,ED50 =塞来昔布(ED50 = 76.09μmol/ kg),ED50 = 46.98-54.45μmol/ kg。同样,与布洛芬(溃疡指数= 20.25)相比,16b,16e,19b和19e的致溃疡性(溃疡指数= 2.79-3.95)明显更少,与塞来昔布(溃疡指数= 2.93)相当。关于抗癌活性,大多数目标衍生物16a-f和19a-f显示出对A-549,MCF-7,HCT-116和PC-3癌细胞系的良好活性。此外,这些衍生物针对F180成纤维细胞进行了研究,以研究其选择性指数。具有内部肟19b和19e的氨磺酰基衍生物是对所有使用过的细胞系最有效的衍生物,尤其是PC-3(分别为IC50 = 1.48和0.33 µM),对PC-3的选择性分别是F180成纤维细胞的11.75和39.4倍。对19b和19e的机理研究表明,这两种化合物在G2 / M期将细胞周期停滞了32.16和39.95倍,将Bax表达上调了6.83和14.52倍,并将基因Bcl-2的表达下调了0.57和。分别是0.36倍 同样,19b和19e是p38MAPK的良好抑制剂(0。与PC-3对照细胞相比,19b为65,19e为0.58)和VEGFR-2(19b为0.39,19e为0.54)。所有化合物16a-f和19a-f均以缓慢的速率(0.15-3.17%)释放NO,并且四种氨磺酰基衍生物16b,16e,19b和19e是最大的NO释放剂(分别为3.06、2.15、3.17和2.54%)。进行了对接研究以解释16a-f和19a-f与靶酶的相互作用。最终设计的化合物与塞来昔布(ID:3LN1)的对接模式表明,它们的三唑环被用作Y形结构中的核心芳基。关于EGFR抑制,以ID:1M17进行对接。严重的内部肟作为抗癌活性更高,因为它们能够与受体裂口形成额外的HBs。所有化合物16a-f和19a-f均以缓慢的速率(0.15-3.17%)释放NO,并且四种氨磺酰基衍生物16b,16e,19b和19e是最大的NO释放剂(分别为3.06、2.15、3.17和2.54%)。进行了对接研究以解释16a-f和19a-f与目标酶的相互作用。最终设计的化合物与塞来昔布(ID:3LN1)的对接模式表明,它们的三唑环被用作Y形结构中的核心芳基。关于EGFR抑制,以ID:1M17进行对接。严重的内部肟作为抗癌活性更高,因为它们能够与受体裂口形成额外的HBs。所有化合物16a-f和19a-f均以缓慢的速率(0.15-3.17%)释放NO,并且四种氨磺酰基衍生物16b,16e,19b和19e是最大的NO释放剂(分别为3.06、2.15、3.17和2.54%)。进行了对接研究以解释16a-f和19a-f与靶酶的相互作用。最终设计的化合物与塞来昔布(ID:3LN1)的对接模式表明,它们的三唑环被用作Y形结构中的核心芳基。关于EGFR抑制,以ID:1M17进行对接。严重的内部肟作为抗癌活性更高,因为它们能够与受体裂口形成额外的HBs。进行了对接研究以解释16a-f和19a-f与靶酶的相互作用。最终设计的化合物与塞来昔布(ID:3LN1)的对接模式表明,它们的三唑环被用作Y形结构中的核心芳基。关于EGFR抑制,以ID:1M17进行对接。严重的内部肟作为抗癌活性更高,因为它们能够与受体裂口形成额外的HBs。进行了对接研究以解释16a-f和19a-f与目标酶的相互作用。最终设计的化合物与塞来昔布(ID:3LN1)的对接模式表明,它们的三唑环被用作Y形结构中的核心芳基。关于EGFR抑制,以ID:1M17进行对接。严重的内部肟作为抗癌活性更高,因为它们能够与受体裂口形成额外的HBs。


























 京公网安备 11010802027423号
京公网安备 11010802027423号