Computational and Theoretical Chemistry ( IF 2.8 ) Pub Date : 2020-03-12 , DOI: 10.1016/j.comptc.2020.112783 Shuang Zhao , Zhe Zhao , Kaisheng Yao , Hui Liu
|
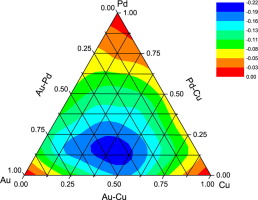
We report a systematic study of ternary PdaCubAuc clusters, with a+b+c=7. The optimized geometries, binding energy per atom, excess binding energy, electrostatic potential surfaces and the physical adsorption behavior of H2 were analyzed by density function theory calculation with TPSSTPSS functional and SDD basis set. PdaCubAuc clusters with more Au atoms exhibit lower binding energy per atom. The average bond distance reduces with increasing Cu content. For PdaCubAuc-H2 clusters, the site precedence for the molecular H2 adsorption follows the order Pd>Cu>Au. The adsorption energy correlates linearly with the elongation of H-H distance.
中文翻译:
Pd a Cu b Au c(a + b + c = 7)团簇的密度泛函研究:几何,电子和H 2物理吸附特性
我们报告了对a + b + c = 7的三元Pd a Cu b Au c团簇的系统研究。通过使用TPSSTPSS函数和SDD基组的密度函数理论计算,分析了H 2的最佳几何构型,每个原子的结合能,过量的结合能,静电势面以及H 2的物理吸附行为。具有更多Au原子的Pd a Cu b Au c团簇每个原子的结合能较低。平均键距随着Cu含量的增加而减小。对于Pd a Cu b Au c -H 2簇,分子H 2吸附的位置优先顺序为Pd> Cu> Au。吸附能与HH距离的延长呈线性关系。

























 京公网安备 11010802027423号
京公网安备 11010802027423号