当前位置:
X-MOL 学术
›
ACS Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Reductive Electrochemical Activation of Molecular Oxygen Catalyzed by an Iron-Tungstate Oxide Capsule: Reactivity Studies Consistent with Compound I Type Oxidants
ACS Catalysis ( IF 12.9 ) Pub Date : 2020-03-19 , DOI: 10.1021/acscatal.0c00897 Marco Bugnola 1 , Kaiji Shen 1 , Eynat Haviv 1 , Ronny Neumann 1
ACS Catalysis ( IF 12.9 ) Pub Date : 2020-03-19 , DOI: 10.1021/acscatal.0c00897 Marco Bugnola 1 , Kaiji Shen 1 , Eynat Haviv 1 , Ronny Neumann 1
Affiliation
|
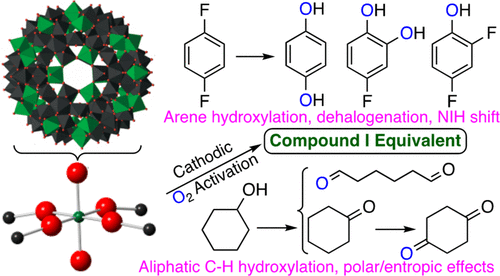
The reductive activation of molecular oxygen catalyzed by iron-based enzymes toward its use as an oxygen donor is paradigmatic for oxygen transfer reactions in nature. Mechanistic studies on these enzymes and related biomimetic coordination compounds designed to form reactive intermediates, almost invariably using various “shunt” pathways, have shown that high-valent Fe(V)═O and the formally isoelectronic Fe(IV)═O porphyrin cation radical intermediates are often thought to be the active species in alkane and arene hydroxylation and alkene epoxidation reactions. Although this four decade long research effort has yielded a massive amount of spectroscopic data, reactivity studies, and a detailed, but still incomplete, mechanistic understanding, the actual reductive activation of molecular oxygen coupled with efficient catalytic transformations has rarely been experimentally studied. Recently, we found that a completely inorganic iron–tungsten oxide capsule with a keplerate structure, noted as {Fe30W72}, is an effective electrocatalyst for the cathodic activation of molecular oxygen in water leading to the oxidation of light alkanes and alkenes. The present report deals with extensive reactivity studies of these {Fe30W72} electrocatalytic reactions showing (1) arene hydroxylation including kinetic isotope effects and migration of the ipso substituent to the adjacent carbon atom (“NIH shift”); (2) a high kinetic isotope effect for alkyl C—H bond activation; (3) dealkylation of alkylamines and alkylsulfides; (4) desaturation reactions; (5) retention of stereochemistry in cis-alkene epoxidation; and (6) unusual regioselectivity in the oxidation of cyclic and acyclic ketones, alcohols, and carboxylic acids where reactivity is not correlated to the bond disassociation energy; the regioselectivity obtained is attributable to polar effects and/or entropic contributions. Collectively these results also support the conclusion that the active intermediate species formed in the catalytic cycle is consistent with a compound I type oxidant. The activity of {Fe30W72} in cathodic aerobic oxidation reactions shows it to be an inorganic functional analogue of iron-based monooxygenases.
中文翻译:

钨酸铁氧化物胶囊催化的分子氧的还原电化学活化:与化合物I型氧化剂一致的反应性研究
铁基酶催化的分子氧的还原活化趋向于用作氧供体是自然界中氧转移反应的典范。对这些酶和相关仿生配位化合物(几乎总是使用各种“分流”途径)旨在形成反应性中间体的机理研究表明,高价Fe(V)═O和形式上等电的Fe(IV)═O卟啉阳离子自由基通常认为中间体是烷烃和芳烃羟基化和烯烃环氧化反应中的活性物质。尽管这四年的长期研究工作已产生了大量的光谱数据,反应性研究,以及对机械机理的详尽但仍不完整的理解,分子氧的实际还原活化结合有效的催化转化很少进行实验研究。最近,我们发现了一个完全无机的氧化钨-钨胶囊,具有keplerate结构,记为{Fe30 W 72 }是一种有效的电催化剂,用于水中的分子氧的阴极活化,从而导致轻质烷烃和烯烃的氧化。本报告对这些{Fe 30 W 72 }电催化反应进行了广泛的反应性研究,结果表明:(1)芳烃羟基化反应,包括动力学同位素效应和ipso取代基向相邻碳原子的迁移(“ NIH位移”);(2)对于烷基CH键活化的高动力学同位素效应;(3)烷基胺和烷基硫化物的脱烷基;(4)去饱和反应;(5)在顺式中保留立体化学烯烃环氧化 (6)在环状和非环状酮,醇和羧酸的氧化反应中,区域反应选择性高,而反应性与键解离能无关。获得的区域选择性归因于极性效应和/或熵贡献。这些结果共同也支持以下结论:在催化循环中形成的活性中间体与化合物I型氧化剂一致。{Fe 30 W 72 }在阴极需氧氧化反应中的活性表明它是铁基单加氧酶的无机功能类似物。
更新日期:2020-03-20
中文翻译:
钨酸铁氧化物胶囊催化的分子氧的还原电化学活化:与化合物I型氧化剂一致的反应性研究
铁基酶催化的分子氧的还原活化趋向于用作氧供体是自然界中氧转移反应的典范。对这些酶和相关仿生配位化合物(几乎总是使用各种“分流”途径)旨在形成反应性中间体的机理研究表明,高价Fe(V)═O和形式上等电的Fe(IV)═O卟啉阳离子自由基通常认为中间体是烷烃和芳烃羟基化和烯烃环氧化反应中的活性物质。尽管这四年的长期研究工作已产生了大量的光谱数据,反应性研究,以及对机械机理的详尽但仍不完整的理解,分子氧的实际还原活化结合有效的催化转化很少进行实验研究。最近,我们发现了一个完全无机的氧化钨-钨胶囊,具有keplerate结构,记为{Fe30 W 72 }是一种有效的电催化剂,用于水中的分子氧的阴极活化,从而导致轻质烷烃和烯烃的氧化。本报告对这些{Fe 30 W 72 }电催化反应进行了广泛的反应性研究,结果表明:(1)芳烃羟基化反应,包括动力学同位素效应和ipso取代基向相邻碳原子的迁移(“ NIH位移”);(2)对于烷基CH键活化的高动力学同位素效应;(3)烷基胺和烷基硫化物的脱烷基;(4)去饱和反应;(5)在顺式中保留立体化学烯烃环氧化 (6)在环状和非环状酮,醇和羧酸的氧化反应中,区域反应选择性高,而反应性与键解离能无关。获得的区域选择性归因于极性效应和/或熵贡献。这些结果共同也支持以下结论:在催化循环中形成的活性中间体与化合物I型氧化剂一致。{Fe 30 W 72 }在阴极需氧氧化反应中的活性表明它是铁基单加氧酶的无机功能类似物。


























 京公网安备 11010802027423号
京公网安备 11010802027423号